血液透析患者第Ⅲ相比較臨床試験1,2)- セベラマー塩酸塩無作為化非盲検実薬対照比較試験:非劣性試験 -
1)承認時評価資料:血液透析患者を対象とした第Ⅲ相比較臨床試験
2)Koiwa F, et al.: Nephrology(Carlton). 2017; 22: 293-300.(本試験はキッセイ薬品工業株式会社の支援により実施された)
血清リン濃度の推移と変化量(主要評価項目)
最終評価時の血清リン濃度におけるセベラマー塩酸塩群に対するピートル群の調整済み※1平均の差[95%信頼区間]は-0.34mg/dL[-0.63,
-0.05:p=0.020(0週時の血清リン濃度を共変量とした共分散分析)]であり、セベラマー塩酸塩に対するピートルの非劣性が検証されました。
※1 0週時の血清リン濃度で調整
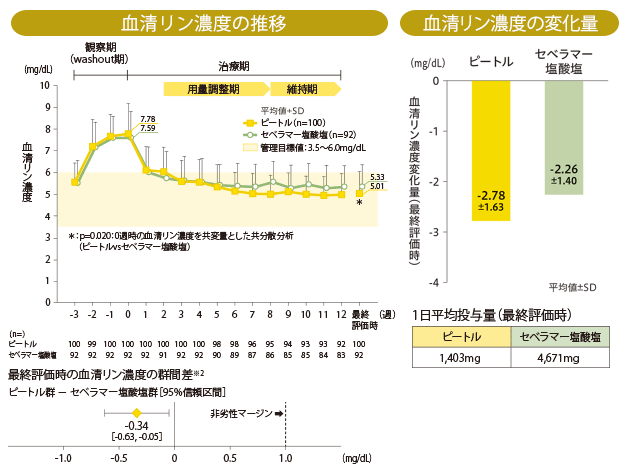
※2 投与群を固定効果、0週時の血清リン濃度を共変量とした共分散分析
【試験概要】
- 目的
- 高リン血症を有する血液透析患者を対象に、ピートルを12週間投与したときの有効性についてセベラマー塩酸塩に対する非劣性の検証および安全性の比較を行う。
- 対象
- 高リン血症を有する血液透析中の慢性腎不全患者213例(PPS※3:192例)
- 方法
-
3週間の観察期間(washout期間)後、ピートルチュアブル錠(750~3,000mg/日;250mg錠使用)またはセベラマー塩酸塩(3~9g/日;250mg錠使用)を1日3回、食直前に用量調整(2~8週)を行いながら、12週間経口投与した。
なお、用量調整は血清リン濃度(目標値:3.5~6.0mg/dL)および忍容性を勘案しながら以下の基準で行った。
・ピートル群:開始用量は750mg/日とし、1回の増減量は750mg/日
・セベラマー塩酸塩群:開始用量は3g/日または6g/日とし、1回の増減量は0.75g/日または1.5g/日
- 試験デザイン
- 無作為化非盲検実薬対照並行群間比較試験(セベラマー塩酸塩に対する非劣性検証試験)。
- 有効性評価項目
- 主要評価項目:血清リン濃度(推移、最終評価時における血清リン濃度、変化量、目標値〈3.5〜6.0mg/dL〉の達成率など)
副次評価項目:補正血清カルシウム濃度、血清intact-PTH濃度など
- 安全性評価項目
- 副作用の発現状況、臨床検査(血液生化学的検査、重炭酸イオン濃度他)など
- 解析計画
-
有効性に関する主要評価項目は血清リン濃度、主要評価変数は最終評価時における血清リン濃度とした。主要評価変数は、群を固定効果、0週時の血清リン濃度を共変量とした共分散分析を行い、対照薬に対する非劣性を検証した。群間差(ピートル群−セベラマー塩酸塩群)の調整済み平均とその両側95%信頼区間を算出した。非劣性マージンを1.0mg/dLとし、両側95%信頼区間上限が非劣性マージン以下の場合、非劣性が検証されたものとした。その他、血清リン濃度について、群ごとに各評価時期における測定値および0週時からの変化量の要約統計量(例数、平均値、SDなど)、目標値達成率を算出した。副次評価項目は、群ごとに各評価時期における測定値の要約統計量を算出した。また、1日平均投与量を算出した。
安全性について、副作用は治験薬投与以降に発現したものを解析対象とし、群ごとに発現件数、発現例数および発現率を、臨床検査は群ごとに各評価時期における測定値の要約統計量を求めた。
| ※3 PPS(Per Protocol Set) : |
有効性解析対象集団。FASから、不適格例、中止例、逸脱例の項目ごとにPPSから除外することが定められた症例を除外した集団。 |
- 7.用法及び用量に関連する注意(抜粋)
- 7.2増量を行う場合は、増量幅を鉄として1日あたりの用量で750mgまでとし、1週間以上の間隔をあけて行うこと。
- 8.重要な基本的注意(抜粋)
- 8.1本剤は、定期的に血清リン、血清カルシウム及び血清PTH濃度を測定しながら投与すること。血清リン、血清カルシウム及び血清PTH濃度の管理目標値及び測定頻度は、学会のガイドライン等、最新の情報を参考にすること。低カルシウム血症の発現あるいは悪化がみられた場合には、活性型ビタミンD製剤やカルシウム製剤の投与を考慮し、カルシウム受容体作動薬が使用されている場合には、カルシウム受容体作動薬の減量等も考慮すること。また、二次性副甲状腺機能亢進症の発現あるいは悪化がみられた場合には、活性型ビタミンD製剤、カルシウム製剤、カルシウム受容体作動薬の投与あるいは他の適切な治療法を考慮すること。
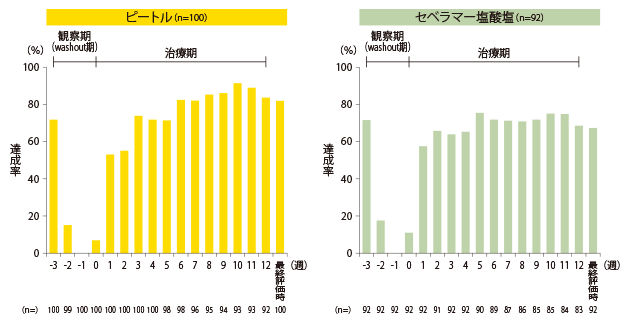
血清リン濃度の目標値達成率(主要評価項目)
ピートル群における血清リン濃度の管理目標値(CKD-MBDの診療ガイドライン:3.5~6.0mg/dL)達成率は82.0%でした(最終評価時)。
【試験概要】
- 目的
- 高リン血症を有する血液透析患者を対象に、ピートルを12週間投与したときの有効性についてセベラマー塩酸塩に対する非劣性の検証および安全性の比較を行う。
- 対象
- 高リン血症を有する血液透析中の慢性腎不全患者213例(PPS※:192例)
- 方法
-
3週間の観察期間(washout期間)後、ピートルチュアブル錠(750~3,000mg/日;250mg錠使用)またはセベラマー塩酸塩(3~9g/日;250mg錠使用)を1日3回、食直前に用量調整(2~8週)を行いながら、12週間経口投与した。
なお、用量調整は血清リン濃度(目標値:3.5~6.0mg/dL)および忍容性を勘案しながら以下の基準で行った。
・ピートル群:開始用量は750mg/日とし、1回の増減量は750mg/日
・セベラマー塩酸塩群:開始用量は3g/日または6g/日とし、1回の増減量は0.75g/日または1.5g/日
- 試験デザイン
- 無作為化非盲検実薬対照並行群間比較試験(セベラマー塩酸塩に対する非劣性検証試験)。
- 有効性評価項目
- 主要評価項目:血清リン濃度(推移、最終評価時における血清リン濃度、変化量、目標値〈3.5〜6.0mg/dL〉の達成率など)
副次評価項目:補正血清カルシウム濃度、血清intact-PTH濃度など
- 安全性評価項目
- 副作用の発現状況、臨床検査(血液生化学的検査、重炭酸イオン濃度他)など
- 解析計画
-
有効性に関する主要評価項目は血清リン濃度、主要評価変数は最終評価時における血清リン濃度とした。主要評価変数は、群を固定効果、0週時の血清リン濃度を共変量とした共分散分析を行い、対照薬に対する非劣性を検証した。群間差(ピートル群−セベラマー塩酸塩群)の調整済み平均とその両側95%信頼区間を算出した。非劣性マージンを1.0mg/dLとし、両側95%信頼区間上限が非劣性マージン以下の場合、非劣性が検証されたものとした。その他、血清リン濃度について、群ごとに各評価時期における測定値および0週時からの変化量の要約統計量(例数、平均値、SDなど)、目標値達成率を算出した。副次評価項目は、群ごとに各評価時期における測定値の要約統計量を算出した。また、1日平均投与量を算出した。
安全性について、副作用は治験薬投与以降に発現したものを解析対象とし、群ごとに発現件数、発現例数および発現率を、臨床検査は群ごとに各評価時期における測定値の要約統計量を求めた。
| ※PPS(Per Protocol Set) : |
有効性解析対象集団。FASから、不適格例、中止例、逸脱例の項目ごとにPPSから除外することが定められた症例を除外した集団。 |
- 7.用法及び用量に関連する注意(抜粋)
- 7.2増量を行う場合は、増量幅を鉄として1日あたりの用量で750mgまでとし、1週間以上の間隔をあけて行うこと。
- 8.重要な基本的注意(抜粋)
- 8.1本剤は、定期的に血清リン、血清カルシウム及び血清PTH濃度を測定しながら投与すること。血清リン、血清カルシウム及び血清PTH濃度の管理目標値及び測定頻度は、学会のガイドライン等、最新の情報を参考にすること。低カルシウム血症の発現あるいは悪化がみられた場合には、活性型ビタミンD製剤やカルシウム製剤の投与を考慮し、カルシウム受容体作動薬が使用されている場合には、カルシウム受容体作動薬の減量等も考慮すること。また、二次性副甲状腺機能亢進症の発現あるいは悪化がみられた場合には、活性型ビタミンD製剤、カルシウム製剤、カルシウム受容体作動薬の投与あるいは他の適切な治療法を考慮すること。
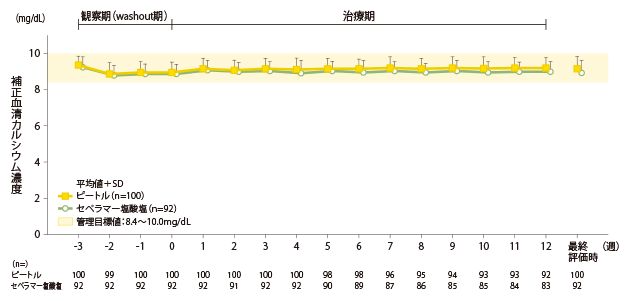
補正血清カルシウム濃度の推移(副次評価項目)
ピートル群の補正血清カルシウム濃度は、0週時8.95±0.58mg/dL、最終評価時9.14±0.68mg/dLと管理目標値(CKD-MBDの診療ガイドライン:8.4~10.0mg/dL)の範囲内でした。
【試験概要】
- 目的
- 高リン血症を有する血液透析患者を対象に、ピートルを12週間投与したときの有効性についてセベラマー塩酸塩に対する非劣性の検証および安全性の比較を行う。
- 対象
- 高リン血症を有する血液透析中の慢性腎不全患者213例(PPS※:192例)
- 方法
-
3週間の観察期間(washout期間)後、ピートルチュアブル錠(750~3,000mg/日;250mg錠使用)またはセベラマー塩酸塩(3~9g/日;250mg錠使用)を1日3回、食直前に用量調整(2~8週)を行いながら、12週間経口投与した。
なお、用量調整は血清リン濃度(目標値:3.5~6.0mg/dL)および忍容性を勘案しながら以下の基準で行った。
・ピートル群:開始用量は750mg/日とし、1回の増減量は750mg/日
・セベラマー塩酸塩群:開始用量は3g/日または6g/日とし、1回の増減量は0.75g/日または1.5g/日
- 試験デザイン
- 無作為化非盲検実薬対照並行群間比較試験(セベラマー塩酸塩に対する非劣性検証試験)。
- 有効性評価項目
- 主要評価項目:血清リン濃度(推移、最終評価時における血清リン濃度、変化量、目標値〈3.5〜6.0mg/dL〉の達成率など)
副次評価項目:補正血清カルシウム濃度、血清intact-PTH濃度など
- 安全性評価項目
- 副作用の発現状況、臨床検査(血液生化学的検査、重炭酸イオン濃度他)など
- 解析計画
-
有効性に関する主要評価項目は血清リン濃度、主要評価変数は最終評価時における血清リン濃度とした。主要評価変数は、群を固定効果、0週時の血清リン濃度を共変量とした共分散分析を行い、対照薬に対する非劣性を検証した。群間差(ピートル群−セベラマー塩酸塩群)の調整済み平均とその両側95%信頼区間を算出した。非劣性マージンを1.0mg/dLとし、両側95%信頼区間上限が非劣性マージン以下の場合、非劣性が検証されたものとした。その他、血清リン濃度について、群ごとに各評価時期における測定値および0週時からの変化量の要約統計量(例数、平均値、SDなど)、目標値達成率を算出した。副次評価項目は、群ごとに各評価時期における測定値の要約統計量を算出した。また、1日平均投与量を算出した。
安全性について、副作用は治験薬投与以降に発現したものを解析対象とし、群ごとに発現件数、発現例数および発現率を、臨床検査は群ごとに各評価時期における測定値の要約統計量を求めた。
| ※PPS(Per Protocol Set) : |
有効性解析対象集団。FASから、不適格例、中止例、逸脱例の項目ごとにPPSから除外することが定められた症例を除外した集団。 |
- 7.用法及び用量に関連する注意(抜粋)
- 7.2増量を行う場合は、増量幅を鉄として1日あたりの用量で750mgまでとし、1週間以上の間隔をあけて行うこと。
- 8.重要な基本的注意(抜粋)
- 8.1本剤は、定期的に血清リン、血清カルシウム及び血清PTH濃度を測定しながら投与すること。血清リン、血清カルシウム及び血清PTH濃度の管理目標値及び測定頻度は、学会のガイドライン等、最新の情報を参考にすること。低カルシウム血症の発現あるいは悪化がみられた場合には、活性型ビタミンD製剤やカルシウム製剤の投与を考慮し、カルシウム受容体作動薬が使用されている場合には、カルシウム受容体作動薬の減量等も考慮すること。また、二次性副甲状腺機能亢進症の発現あるいは悪化がみられた場合には、活性型ビタミンD製剤、カルシウム製剤、カルシウム受容体作動薬の投与あるいは他の適切な治療法を考慮すること。
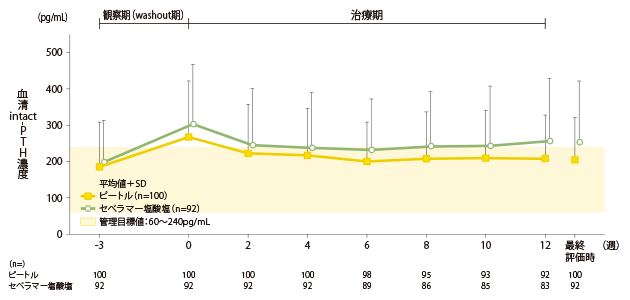
血清intact-PTH濃度の推移(副次評価項目)
ピートル群の血清intact-PTH濃度は、-3週時185.1±123.9pg/mL、0週時267.4±153.6pg/mL、最終評価時203.6±117.1pg/mLでした。
【試験概要】
- 目的
- 高リン血症を有する血液透析患者を対象に、ピートルを12週間投与したときの有効性についてセベラマー塩酸塩に対する非劣性の検証および安全性の比較を行う。
- 対象
- 高リン血症を有する血液透析中の慢性腎不全患者213例(PPS※:192例)
- 方法
-
3週間の観察期間(washout期間)後、ピートルチュアブル錠(750~3,000mg/日;250mg錠使用)またはセベラマー塩酸塩(3~9g/日;250mg錠使用)を1日3回、食直前に用量調整(2~8週)を行いながら、12週間経口投与した。
なお、用量調整は血清リン濃度(目標値:3.5~6.0mg/dL)および忍容性を勘案しながら以下の基準で行った。
・ピートル群:開始用量は750mg/日とし、1回の増減量は750mg/日
・セベラマー塩酸塩群:開始用量は3g/日または6g/日とし、1回の増減量は0.75g/日または1.5g/日
- 試験デザイン
- 無作為化非盲検実薬対照並行群間比較試験(セベラマー塩酸塩に対する非劣性検証試験)。
- 有効性評価項目
- 主要評価項目:血清リン濃度(推移、最終評価時における血清リン濃度、変化量、目標値〈3.5〜6.0mg/dL〉の達成率など)
副次評価項目:補正血清カルシウム濃度、血清intact-PTH濃度など
- 安全性評価項目
- 副作用の発現状況、臨床検査(血液生化学的検査、重炭酸イオン濃度他)など
- 解析計画
-
有効性に関する主要評価項目は血清リン濃度、主要評価変数は最終評価時における血清リン濃度とした。主要評価変数は、群を固定効果、0週時の血清リン濃度を共変量とした共分散分析を行い、対照薬に対する非劣性を検証した。群間差(ピートル群−セベラマー塩酸塩群)の調整済み平均とその両側95%信頼区間を算出した。非劣性マージンを1.0mg/dLとし、両側95%信頼区間上限が非劣性マージン以下の場合、非劣性が検証されたものとした。その他、血清リン濃度について、群ごとに各評価時期における測定値および0週時からの変化量の要約統計量(例数、平均値、SDなど)、目標値達成率を算出した。副次評価項目は、群ごとに各評価時期における測定値の要約統計量を算出した。また、1日平均投与量を算出した。
安全性について、副作用は治験薬投与以降に発現したものを解析対象とし、群ごとに発現件数、発現例数および発現率を、臨床検査は群ごとに各評価時期における測定値の要約統計量を求めた。
| ※PPS(Per Protocol Set) : |
有効性解析対象集団。FASから、不適格例、中止例、逸脱例の項目ごとにPPSから除外することが定められた症例を除外した集団。 |
- 7.用法及び用量に関連する注意(抜粋)
- 7.2増量を行う場合は、増量幅を鉄として1日あたりの用量で750mgまでとし、1週間以上の間隔をあけて行うこと。
- 8.重要な基本的注意(抜粋)
- 8.1本剤は、定期的に血清リン、血清カルシウム及び血清PTH濃度を測定しながら投与すること。血清リン、血清カルシウム及び血清PTH濃度の管理目標値及び測定頻度は、学会のガイドライン等、最新の情報を参考にすること。低カルシウム血症の発現あるいは悪化がみられた場合には、活性型ビタミンD製剤やカルシウム製剤の投与を考慮し、カルシウム受容体作動薬が使用されている場合には、カルシウム受容体作動薬の減量等も考慮すること。また、二次性副甲状腺機能亢進症の発現あるいは悪化がみられた場合には、活性型ビタミンD製剤、カルシウム製剤、カルシウム受容体作動薬の投与あるいは他の適切な治療法を考慮すること。
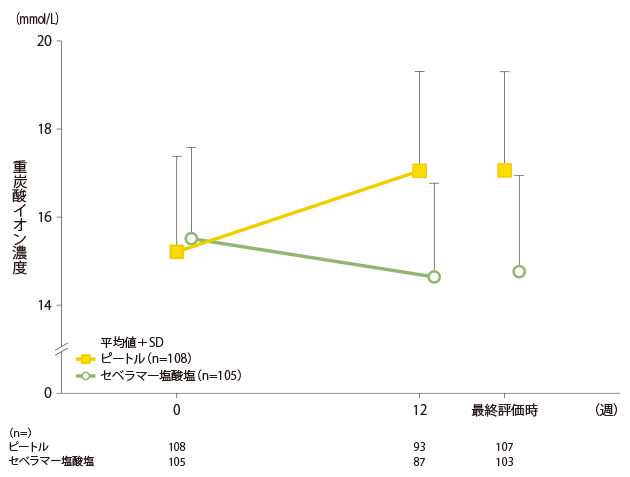
安全性:重炭酸イオン濃度への影響
ピートル群の重炭酸イオン濃度は以下のとおりでした。
【試験概要】
- 目的
- 高リン血症を有する血液透析患者を対象に、ピートルを12週間投与したときの有効性についてセベラマー塩酸塩に対する非劣性の検証および安全性の比較を行う。
- 対象
- 高リン血症を有する血液透析中の慢性腎不全患者213例(PPS※:192例)
- 方法
-
3週間の観察期間(washout期間)後、ピートルチュアブル錠(750~3,000mg/日;250mg錠使用)またはセベラマー塩酸塩(3~9g/日;250mg錠使用)を1日3回、食直前に用量調整(2~8週)を行いながら、12週間経口投与した。
なお、用量調整は血清リン濃度(目標値:3.5~6.0mg/dL)および忍容性を勘案しながら以下の基準で行った。
・ピートル群:開始用量は750mg/日とし、1回の増減量は750mg/日
・セベラマー塩酸塩群:開始用量は3g/日または6g/日とし、1回の増減量は0.75g/日または1.5g/日
- 試験デザイン
- 無作為化非盲検実薬対照並行群間比較試験(セベラマー塩酸塩に対する非劣性検証試験)。
- 有効性評価項目
- 主要評価項目:血清リン濃度(推移、最終評価時における血清リン濃度、変化量、目標値〈3.5〜6.0mg/dL〉の達成率など)
副次評価項目:補正血清カルシウム濃度、血清intact-PTH濃度など
- 安全性評価項目
- 副作用の発現状況、臨床検査(血液生化学的検査、重炭酸イオン濃度他)など
- 解析計画
-
有効性に関する主要評価項目は血清リン濃度、主要評価変数は最終評価時における血清リン濃度とした。主要評価変数は、群を固定効果、0週時の血清リン濃度を共変量とした共分散分析を行い、対照薬に対する非劣性を検証した。群間差(ピートル群−セベラマー塩酸塩群)の調整済み平均とその両側95%信頼区間を算出した。非劣性マージンを1.0mg/dLとし、両側95%信頼区間上限が非劣性マージン以下の場合、非劣性が検証されたものとした。その他、血清リン濃度について、群ごとに各評価時期における測定値および0週時からの変化量の要約統計量(例数、平均値、SDなど)、目標値達成率を算出した。副次評価項目は、群ごとに各評価時期における測定値の要約統計量を算出した。また、1日平均投与量を算出した。
安全性について、副作用は治験薬投与以降に発現したものを解析対象とし、群ごとに発現件数、発現例数および発現率を、臨床検査は群ごとに各評価時期における測定値の要約統計量を求めた。
| ※PPS(Per Protocol Set) : |
有効性解析対象集団。FASから、不適格例、中止例、逸脱例の項目ごとにPPSから除外することが定められた症例を除外した集団。 |
- 7.用法及び用量に関連する注意(抜粋)
- 7.2増量を行う場合は、増量幅を鉄として1日あたりの用量で750mgまでとし、1週間以上の間隔をあけて行うこと。
- 8.重要な基本的注意(抜粋)
- 8.1本剤は、定期的に血清リン、血清カルシウム及び血清PTH濃度を測定しながら投与すること。血清リン、血清カルシウム及び血清PTH濃度の管理目標値及び測定頻度は、学会のガイドライン等、最新の情報を参考にすること。低カルシウム血症の発現あるいは悪化がみられた場合には、活性型ビタミンD製剤やカルシウム製剤の投与を考慮し、カルシウム受容体作動薬が使用されている場合には、カルシウム受容体作動薬の減量等も考慮すること。また、二次性副甲状腺機能亢進症の発現あるいは悪化がみられた場合には、活性型ビタミンD製剤、カルシウム製剤、カルシウム受容体作動薬の投与あるいは他の適切な治療法を考慮すること。
安全性:副作用
副作用発現率はピートル群で26.9%(29/108例)、セベラマー塩酸塩群26.7%(28/105例)でした。ピートル群の主な副作用は下痢21.3%(23例)、セベラマー塩酸塩群では便秘18.1%(19例)でした。
なお、重篤な副作用として、ピートル群において急性肺水腫が1例、うっ血性心不全が1例、セベラマー塩酸塩群において憩室炎が1例に認められました。投与中止に至った副作用として、ピートル群において下痢が4例、胃腸炎が1例、ヘモグロビン増加が1例、セベラマー塩酸塩群において便秘が3例、腹痛が1例、下痢が1例、悪心が1例、食欲減退が1例、腹部不快感が1例に認められました。



 ピートル
ピートル