


 イセルティ
イセルティ
国内第Ⅲ相臨床試験(KLH2302)1)
1)社内資料(承認時評価資料):国内第Ⅲ相臨床試験(KLH2302)(CTD2.7.6.13)
目的
過多月経及び疼痛症状を有する子宮筋腫患者を対象として、イセルティ®200mgを1日1回12週間経口投与したときの過多月経及び疼痛症状に対する有効性について、プラセボに対する優越性を二重盲検法により検証する。
試験デザイン
プラセボを対照とした多施設共同無作為化二重盲検並行群間比較試験
対象
20歳以上の閉経前の過多月経及び疼痛症状を有する子宮筋腫患者89例(イセルティ®群48例、プラセボ群41例)
主な選択基準
- 経腟超音波、腹部超音波、MRI、CT、腹腔鏡検査のいずれかによって子宮筋腫と診断されている患者
- スクリーニング期開始時・治療期開始時の経膣超音波検査で、以下の条件をすべて満たす筋腫核を1つ以上有していると確認された患者
- 最長径が3cm以上、石灰化を伴っていない、外科的治療を受けていない
- 治療期開始直前の月経周期1回分のPBACスコアの合計点が120点以上で、過多月経と診断された患者
- スクリーニング期開始直前・前観察期開始直前・治療期開始直前に正常な月経周期(25~38日)を認め、かつ3日以上の連続的な出血を伴う月経が当該月経周期中に確認されている患者
- 治療期開始直前の月経周期1回分の子宮筋腫の疼痛症状に対するNRSスコア最大値が4以上である患者
方法
試験は、スクリーニング期、単盲検下でプラセボを投与する前観察期、二重盲検下で治験薬を投与する治療期12週間及び後観察期4週間から構成された。
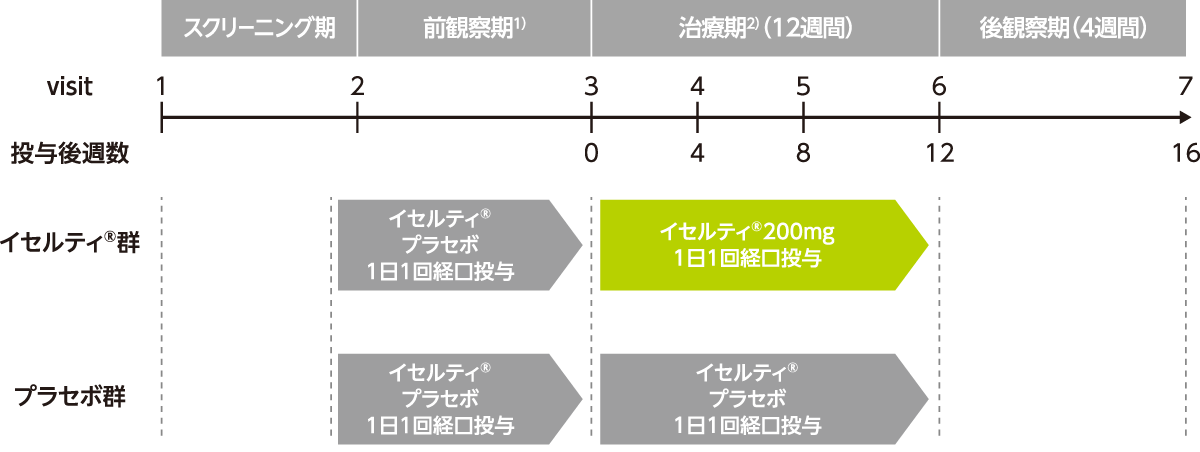
- 1)スクリーニング期開始後1回目の月経開始1~5日目の来院を前観察期開始時とする。ただし、前観察期開始時の選択基準を満たしているもののスクリーニング期開始後1回目の月経開始1~5日目に来院できない場合は、2回目の月経開始1~5日目の来院を前観察期開始時とすることも可とする。
- 2)スクリーニング期開始後2回目の月経開始1~5日目の来院を治療期開始時とする。ただし、スクリーニング期開始後2回目の月経開始1~5日目に前観察期を開始した場合は、3回目の月経開始1~5日目の来院を治療期開始時とする。治療薬投与終了日+7日以内に実施された観察・検査項目のうち、規定の来院で最も遅い時点のデータを治療期最終評価時データとする。
- イセルティ®群:前観察期にプラセボを、治療期にイセルティ®200mgを1日1回経口投与する。
初回投与は月経1~5日目に実施する。
- プラセボ群:前観察期・治療期にプラセボを1日1回経口投与する。
初回投与は月経1~5日目に実施する。
評価項目
有効性評価項目
主要評価項目
- 治験薬投与6週後から12週後までのPBACスコア*1の合計点が10点未満である患者割合(検証的解析項目)
- 治験薬投与終了前28日間における疼痛症状に対するNumerical rating scale(NRS)スコア*2最大値が1以下である患者割合(検証的解析項目)
副次評価項目
- PBACスコアの合計点が10点未満である患者割合(治験薬投与2週後から6週後まで及び投与終了前6週間)
- NRSスコア最大値の推移及びベースラインからの変化量
- 28日間ごとの疼痛症状に対するNRSスコア最大値が1以下である患者割合
- 28日間ごとの無症状日数(疼痛症状に対するNRSスコア0の日数)の割合のベースラインからの変化量
- 評価時点ごとの血中ヘモグロビン値の推移、ベースラインからの変化量
- 評価時点ごとのUFS-QOLスコア*3のベースラインからの変化量
- 治療期最終評価時のPGIC*4の全般的症状の割合 など
有効性に関するサブグループ解析
- 疼痛症状に関する主要評価項目(治験薬投与終了前28日間における疼痛症状に対するNRSスコア最大値が1以下である患者割合)におけるベースラインのNRSスコア最大値が4以上7未満/7以上の部分集団ごとの改善例の割合
- ベースラインの血中ヘモグロビン値が12g/dL未満の患者における評価時点ごとの血中ヘモグロビン値の推移及びベースラインからの変化量
安全性評価項目
副作用の発現割合、治験薬最終投与から月経回復までの期間 など
解析計画
有効性評価項目
主要評価項目
以下の過多月経及び疼痛症状に関する主要評価項目がいずれも検証された場合にのみ試験の主要目的が達成されるものとした。
■治験薬投与6週後から12週後までのPBACスコアの合計点が10点未満である患者割合
■治験薬投与終了前28日間における疼痛症状に対するNRSスコア最大値が1以下である患者割合
- 治験薬投与6週後から12週後までのPBACスコアの合計点が10点未満を改善例とし、群ごとに改善例の例数及び割合と両側95%CIを示した。治験薬投与終了前28日間における疼痛症状に対するNRSスコア最大値が1以下を改善例とし、群ごとに改善例の例数及び割合と両側95%CIを示した。イセルティ®群とプラセボ群の群間差(イセルティ®群-プラセボ群)の点推定値及びその両側95%CIを示し、Fisherの正確検定を用いてプラセボ群に対するイセルティ®群の優越性を検証した。
副次評価項目
- PBACスコアの合計点が10点未満である患者割合は、群間差(イセルティ®群-プラセボ群)の点推定値及びその両側95%CIを示した。
- 28日間ごとの疼痛症状に対するNRSスコア最大値が1以下である患者(改善例)割合について、改善例の割合とその両側95%CIを示し、改善例の割合を図示した。イセルティ®群とプラセボ群の群間差(イセルティ®群-プラセボ群)の点推定値及びその両側95%CIを示した。
- NRSスコア最大値の推移は、要約統計量を示し、平均値及び標準偏差を図示した。ベースラインからの変化量は、要約統計量を示し、平均値及び標準偏差を図示し、イセルティ®群とプラセボ群の群間差(イセルティ®群-プラセボ群)の点推定値及びその両側95%CIを示した。
- 28日間ごとの無症状日数の割合のベースラインからの変化量は、NRSスコア最大値のベースラインからの変化量と同様の解析を行った。
- 評価時点ごとの血中ヘモグロビン値の推移及びベースラインからの変化量の要約統計量を示し、平均値及び標準偏差を図示した。ベースラインからの変化量は群間差(イセルティ®群-プラセボ群)の点推定値及びその両側95%CIを示した。
- UFS-QOLスコアのベースラインからの変化量は、要約統計量を示し、群間差(イセルティ®群-プラセボ群)の点推定値及びその両側95%CIを示した。
- PGICは、全般的症状の割合を示し、2標本Wilcoxon検定を用いて群間比較を行った。
有効性に関するサブグループ解析
- 疼痛症状に関する主要評価項目(治験薬投与終了前28日間における疼痛症状に対するNRSスコア最大値が1以下である患者割合)において、ベースラインのNRSスコア最大値が4以上7未満/7以上の患者別の部分集団ごとの改善例の例数及び割合とその両側95%CIを示した。
- ベースラインの血中ヘモグロビン値を12g/dL未満、12g/dL以上の評価区分に分類した。ベースラインの血中ヘモグロビン値が12g/dL未満の部分集団において血中ヘモグロビン値の推移及びベースラインからの変化量は評価時点ごとの要約統計量を示し、平均値及び標準偏差を図示した。また、ベースラインからの変化量はイセルティ®群とプラセボ群の群間差(イセルティ®群-プラセボ群)の点推定値及びその両側95%CIを示した。
安全性評価項目
- 群ごとに副作用の発現例数及び発現割合を示した。
- 治験薬最終投与から月経回復までの期間の要約統計量を示した。
患者背景
| イセルティ®群(n=48) | プラセボ群(n=41) | 全体(n=89) | |
|---|---|---|---|
| 年齢(歳) | |||
| 平均値±標準偏差 | 41.8±5.4 | 44.4±6.0 | 43.0±5.8 |
| 40歳未満 n(%) | 14(29.2) | 6(14.6) | 20(22.5) |
| 40歳以上 n(%) | 34(70.8) | 35(85.4) | 69(77.5) |
| 体重(kg) | |||
| 平均値±標準偏差 | 60.53±14.49 | 62.20±11.60 | 61.30±13.19 |
| BMI(kg/m2) | |||
| 平均値±標準偏差 | 23.56±4.64 | 24.42±3.92 | 23.96±4.32 |
| 子宮筋腫の種類 n(%) | |||
| 漿膜下筋腫 | 17(35.4) | 16(39.0) | 33(37.1) |
| 筋層内筋腫 | 42(87.5) | 38(92.7) | 80(89.9) |
| 粘膜下筋腫 | 4(8.3) | 2(4.9) | 6(6.7) |
| 子宮頸部筋腫 | 0(0.0) | 1(2.4) | 1(1.1) |
| 子宮筋腫既往期間(年) | |||
| 平均値±標準偏差 | 7.56±6.68 | 7.27±6.56 | 7.43±6.59 |
| 出産経験 n(%) | |||
| なし | 22(45.8) | 20(48.8) | 42(47.2) |
| あり | 26(54.2) | 21(51.2) | 47(52.8) |
| 子宮筋腫に対する薬物治療歴 n(%) | |||
| なし | 31(64.6) | 28(68.3) | 59(66.3) |
| あり | 17(35.4) | 13(31.7) | 30(33.7) |
| 子宮筋腫に対する手術歴 n(%) | |||
| なし | 40(83.3) | 39(95.1) | 79(88.8) |
| あり | 8(16.7) | 2(4.9) | 10(11.2) |
| 筋腫核体積(cm3) | |||
| 平均値±標準偏差 | 66.74±73.27 | 76.18±156.41 | 71.09±118.36 |
| 子宮体積(cm3) | |||
| 平均値±標準偏差 | 204.07±122.11 | 200.41±200.29 | 202.38±161.87 |
| 総PBACスコア*1(点) | |||
| 平均値±標準偏差 | 261.1±154.1 | 348.6±235.7 | 301.4±199.7 |
| 最大NRSスコア*2 | |||
| 平均値±標準偏差 | 6.8±1.9 | 6.2±1.3 | 6.5±1.7 |
| 4以上7未満 n(%) | 22(45.8) | 25(61.0) | 47(52.8) |
| 7以上 n(%) | 26(54.2) | 16(39.0) | 42(47.2) |
| ヘモグロビン値(g/dL) | |||
| 平均値±標準偏差 | 11.90±1.32 | 12.33±1.27 | 12.09±1.31 |
| 12未満 n(%) | 26(54.2) | 18(43.9) | 44(49.4) |
| 12以上 n(%) | 22(45.8) | 23(56.1) | 45(50.6) |
| UFS-QOLスコア*3 Symptom Severity | |||
| 平均値±標準偏差 | 34.70±16.30 | 33.99±18.24 | 34.38±17.12 |
| HRQL Total | |||
| 平均値±標準偏差 | 68.66±22.31 | 71.19±22.54 | 69.83±22.33 |
*1:PBACスコア参照、*2:NRSスコア参照、*3:UFS-QOLスコア参照
評価スコア
- *2:NRSスコア2)
-
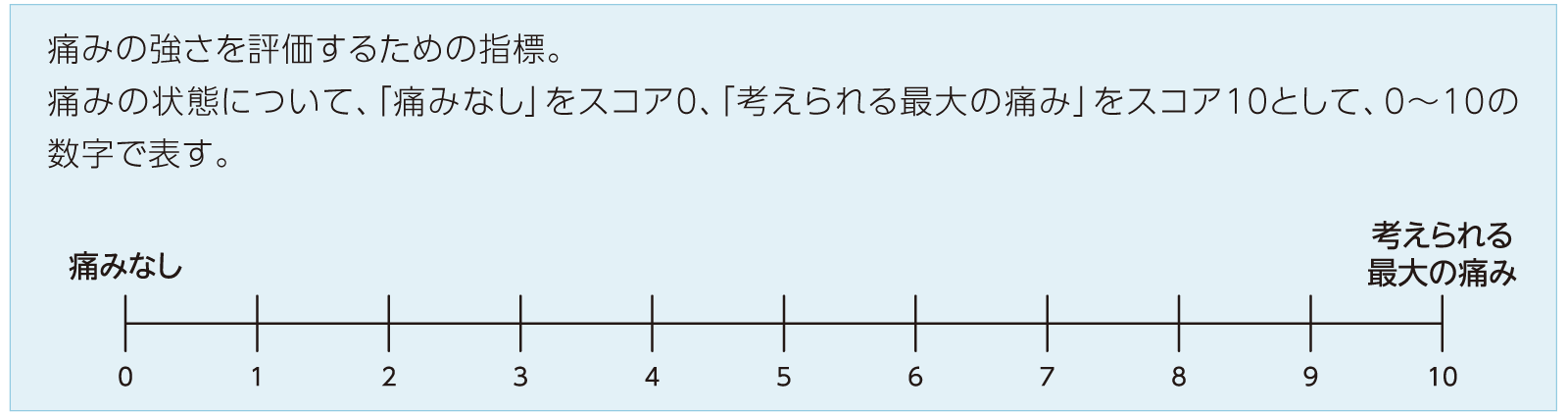
- *3:UFS-QOLスコア3)
-

- *4:PGIC2)
-

2)社内資料(承認時評価資料):有効性評価項目(CTD2.7.3.1.2)
3)Spies JB, et al. Obstet Gynecol. 2002; 99(2): 290-300
3)Spies JB, et al. Obstet Gynecol. 2002; 99(2): 290-300
- 6. 用法及び用量 通常、成人にはリンザゴリクスとして200mgを1日1回経口投与する。なお、初回投与は月経周期1~5日目に行う。
- 7. 用法及び用量に関連する注意(抜粋)
- 7.1治療に際しては妊娠していないことを確認し、必ず月経周期1~5日目より投与を開始すること。
また、治療期間中は非ホルモン性の避妊をさせること。[2.1、9.5参照]
