


試験概要
試験概要
目的
日本人の慢性特発性血小板減少性紫斑病(ITP)患者を対象に、タバリス100~150mgを1日2回、経口投与したときの有効性及び安全性を評価する。
試験デザイン
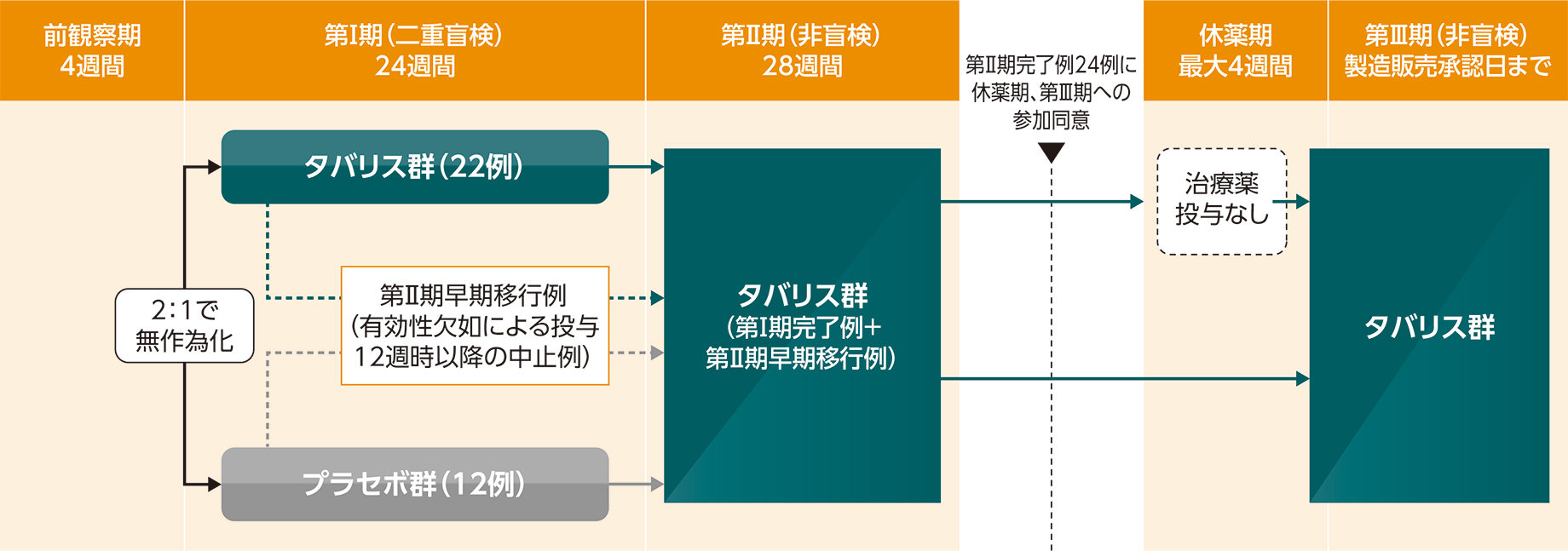
多施設共同、プラセボ対照、無作為化、二重盲検、並行群間(第I期)/非盲検(第II及びIII期)、第III相試験(全国42施設)
対象
慢性ITP患者34例[第I期:タバリス群22例及びプラセボ群12例、第II期:30例(データカットオフ日:2022年5月25日)]
投与方法
[前観察期]治験薬を投与せず、本試験に組み入れる適格性を確認した。[第I期]タバリス群又はプラセボ群に2:1の比率で無作為に割り付け、24週間経口投与した。投与開始時の用法・用量は100mgを1日2回とし、投与4週時以降は血小板数及び忍容性を考慮して150mgを1日2回に増量した。[第II期]第I期を完了した患者及び第II期早期移行例を対象として、タバリスを28週間経口投与した。(1)第I期を完了した患者:投与24週時の来院前まで第I期の治験薬を投与し、投与24週時の来院後から第II期の治験薬の投与を開始した。(2)第II期早期移行患者:早期移行の1日目の来院後から第II期の治験薬の投与を開始した。[休薬期]第II期を完了した患者のうち、休薬期及び第III期への参加の同意が得られた患者を対象に、治験薬を休薬したときの血小板数の推移を検討した。[第III期]タバリスの製造販売承認日まで経口投与を継続した。[第I-III期]治験薬投与期間中は、同日の朝夕の投与は少なくとも8時間は間隔を空け、食事の有無に関わらず可能な限り同じ時間帯に投与した。投与量調整の基準に該当する有害事象が認められた場合は、基準に従って治験薬を調整した。併用可能なITP治療薬として、副腎皮質ステロイド(プレドニゾロン換算で10mg/日以下)、アザチオプリン又はダナゾールのうち1種類を使用可能とし、治験薬投与開始2週間前から第I期中は用法・用量を変更不可とした。第II期及び第III期中は血小板数が50,000/μL以上で安定している患者では減量可能とした。また、血小板数が50,000/μL未満でレスキュー治療が必要な場合はレスキュー薬を使用可能とした。
治験薬の用量調節に使用した基準
治験薬の投与量レベル
| 1日投与量 | 午前 | 午後 |
|---|---|---|
| 300mg/日 | 150mg | 150mg |
| 200mg/日 | 100mg | 100mg |
| 150mg/日 | 150mg | - |
| 100mg/日 | 100mg | - |
レスキュー薬の用法及び用量
| 薬剤 | 用法及び用量 |
|---|---|
| 血小板輸血 | 10~20単位/回 |
| 静注免疫グロブリン製剤 | 400mg/kg/日を連続5日間 |
| 静注メチルプレドニゾロン | 1g/日を連続3日間 |
| 経口デキサメタゾン | 最大40mg/日を1~2日 |
| 経口プレドニゾロン | 最大1mg/kg/日を1~3日 |
評価項目
主要評価項目:第I期におけるStable platelet responseの達成割合
Stable platelet responseは、投与14~24週までの6回の来院のうち4回以上で血小板数が50,000/μL以上と定義し、達成した患者をレスポンダーとした。
副次評価項目
- ・第I期におけるOverall response(投与2~12週までの6回の測定のうち、1回以上で血小板数が50,000/μL以上と定義)の達成割合
- ・血小板数の要約統計量
- ・血小板数の個別推移
- ・血小板数の達成割合
- ・血小板数の奏効率 等
安全性評価項目:有害事象、治験薬との関連性及び副作用の発現状況 等
解析計画
解析対象集団:有効性解析対象集団は、無作為割付けされ、GCP違反、未投薬、第I期移行前中止、不適格の一部の患者及び主要評価項目が得られていない患者を除外した最大の解析対象集団(FAS)とした。
5. 効能又は効果に関連する注意
以下の場合で、診療ガイドライン等の最新の情報を参考に、本剤の投与が適切と判断される患者に投与すること。
- ● 他の治療にて十分な効果が得られない場合、又は忍容性に問題があると考えられる場合
- ● 血小板数、臨床症状からみて出血リスクが高いと考えられる場合
安全性評価は、第I期の「プラセボ対照期」、第I期でタバリス群に割り付けられた患者を対象に第II期のデータも含めて評価した「タバリス長期投与時」、並びに第I期又は第II期のいずれかでタバリスを投与した患者の第III期までのタバリスの全投与期間を評価した「タバリス投与期」を対象とした。
主要評価項目は、Stable platelet responseを達成した患者(レスポンダー)の割合とその群間差について、それぞれ正確な両側95%信頼区間(CI)を算出(それぞれClopper-Pearson法及びChan and Zhang法)し、Fisher's Exact検定を用いてタバリス群のプラセボ群に対する優越性を評価した。なお、有害事象又は有効性の欠如により中止した患者及び投与10~24週時までにレスキュー薬を使用した患者はノンレスポンダーとし、それ以外の患者の欠測は、投与24週時までLast Observation Carried Forward(LOCF)を用いてデータを補完した。
副次評価項目は、それぞれ以下のとおり解析した。
Overall responseの達成割合:達成例数及び割合とその両側95%CIを示す。第I期の評価では、達成割合の群間差とその両側95%CIを示す。
血小板数の要約統計量:血小板数及び血小板数のベースラインからの変化量の要約統計量を示す。中央値及び四分位点について図示する。また、第I期の評価ではStable platelet responseのレスポンダー、ノンレスポンダーごとに部分集団解析を実施し、休薬期の評価では休薬期開始時からの変化量の要約統計量を示す。
血小板数の達成割合:投与52週時における血小板数50,000/μL以上の達成割合について、達成割合とその両側95%CIを示す。
なお、中止又は欠測によりレスポンダーと判定されなかった患者はすべてノンレスポンダーとする。
血小板数の奏効率:治療ライン(セカンドライン、サードライン以降)ごとにタバリス投与期のタバリス投与開始後、投与52週時(第II期)までの評価時点において、1度でも血小板数50,000/μL以上を達成した患者の例数及び割合を示す。
安全性評価項目は、副作用及び特に注目すべき有害事象※、有害事象の発現状況を解析した。
※:出血関連有害事象、胃腸障害、感染症、高血圧、好中球減少症、肝機能障害、血栓症・塞栓症・血栓塞栓症
試験スケジュール
第II期早期移行の基準
以下の基準をすべて満たす患者
以下のいずれかに該当し、「有効性の欠如(効果不十分)※1」と判断され、第I期を中止した患者
- ・投与12週時以降に血小板数が50,000/μL未満※1(投与1日目の血小板数が15,000/μL以上の患者)
- ・投与12週時以降に投与1日目からの血小板数の増加が20,000/μL未満※2(投与1日目の血小板数が15,000/μL未満の患者)
早期移行時あるいは早期移行時の直近の臨床検査にて、白血球数、好中球数、リンパ球数、ヘモグロビン、ALT、AST、総ビリルビン、eGFRの臨床検査値異常が認められなかった患者
早期移行時に、収縮期血圧が140mmHg未満かつ拡張期血圧が90mmHg未満の患者
早期移行時に重大な感染症、インフルエンザなどの急性感染又は活動性の炎症性反応が認められなかった患者
早期移行時の前2週間以内に輸血又は血液製剤を使用していない患者※3
※1:治験薬の有効性が認められず、治験の継続によって、患者に許容できないリスクがあると治験責任医師又は治験分担医師が判断した場合
※2:150mgを1日2回に増量して4週間以上投与した場合、もしくは、忍容性に問題があると考えられたため、150mgを1日2回に増量できない場合
※3:レスキュー薬として使用される静注免疫グロブリン製剤及び血小板輸血は除く

