


 マリゼブ
マリゼブ
臨床成績
1.国際共同後期第Ⅱ相臨床試験(用量設定試験) 1)
1)社内資料(承認時評価資料):国際共同後期第Ⅱ相用量設定試験(P006-00)(2015年9月28日承認、CTD2.7.6.3.1)
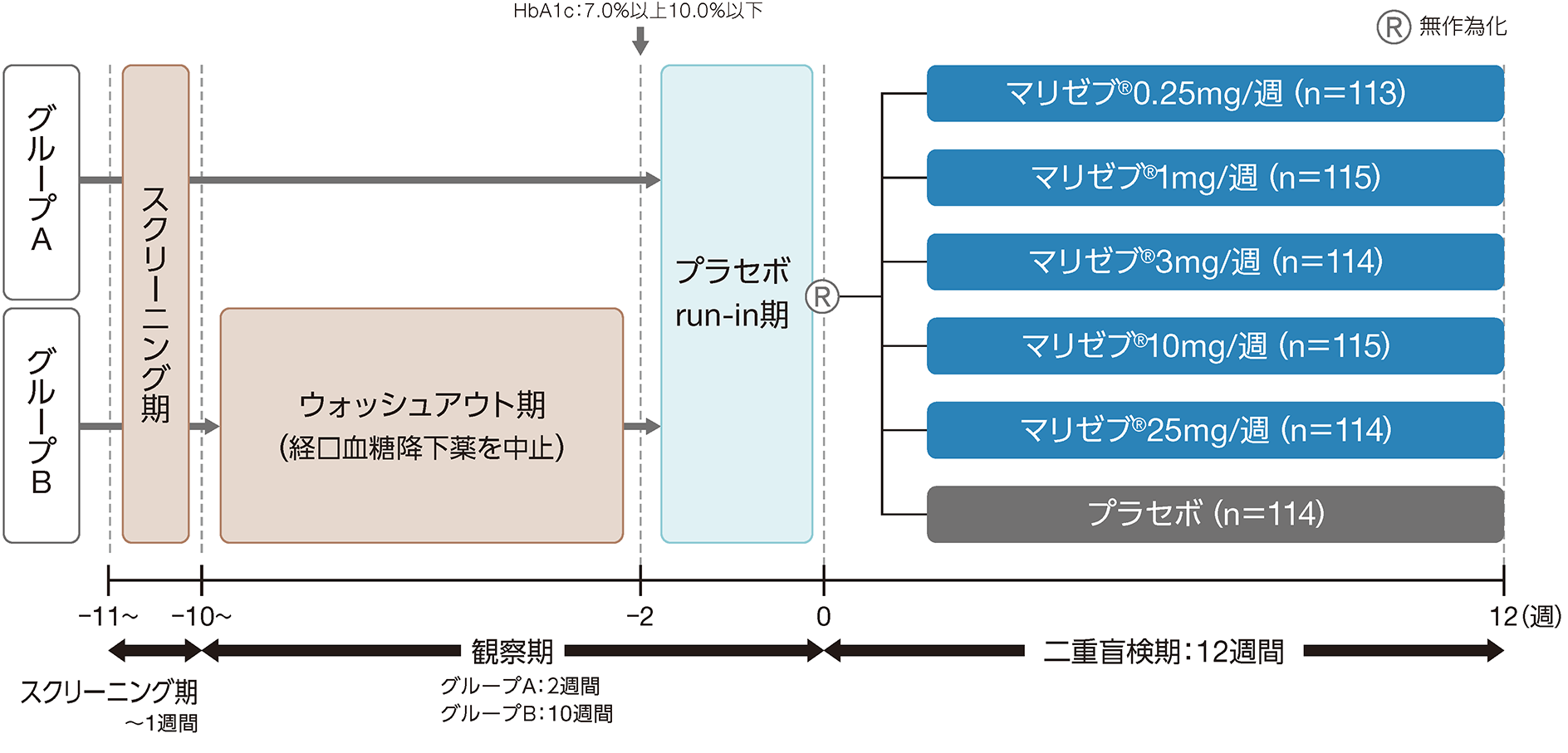
試験概要
紹介する結果には承認用量と異なる成績が含まれていますが、承認時評価資料であるため掲載しています。なお、承認用量(25mg)以外の用量につきましては、安全性に関する結果のみ掲載しています。
- 目的:
- 食事/運動療法で血糖管理が不十分な2型糖尿病患者に対しマリゼブ®の投与12週間後における有効性と安全性及び忍容性を検討する。
- 対象:
- 食事/運動療法で血糖管理不十分な2型糖尿病患者685例(うち日本人127例)
- 血糖降下薬14週間以上未投与でHbA1c7.0%以上10.0%以下の患者(グループA:観察期2週間)
- チアゾリジン薬以外の経口血糖降下薬を単剤もしくは低用量 ※で2剤併用投与中のHbA1c6.5%以上9.0%以下の患者(グループB:観察期10週間)
[FAS *解析対象例数:マリゼブ®0.25mg/週 群113例、1mg/週 群115例、3mg/週 群114例、10mg/週 群115例、25mg/週 群114例、プラセボ群113例]
- 試験デザイン:
- 多施設共同二重盲検無作為化プラセボ対照用量設定試験
- 投与方法:
- プラセボを単盲検下で週1回2週間経口投与した後、マリゼブ®0.25mg~25mgの各投与群またはプラセボ群に無作為に割り付け、二重盲検下でそれぞれ週1回12週間経口投与を行った。投与1日目以降、血糖値が基準に達しない場合はメトホルミンによるレスキュー治療を行った。
- 評価項目:
-
<主要評価項目> 投与12週時のHbA1cのベースライン時(投与0週時)からの変化量(検証的解析項目) <その他の有効性評価項目> 食後2時間血糖値、空腹時血糖値、HbA1c値7.0%未満または6.5%未満達成率、体重、HOMA-β及びレスキュー治療開始までの時間、血漿中薬物濃度及びDPP-4活性阻害率 <安全性評価項目> 有害事象、臨床検査値等 - 解析計画:
-
- 主要評価項目は制約つき経時データ解析の方法(以下、cLDAモデル)を用い、線形対比を用いた下降手順の傾向性検定により評価した。
- その他の有効性評価項目は主要評価項目に対して用いた同様のcLDAモデルを用いて解析を行った。
- 安全性評価項目は事前に規定した特に関心のある有害事象を「Tier 1」の事象(症候性低血糖)とし、群間比較に関する統計的検定のp値及び95%信頼区間を算出した。その他の有害事象(「Tier 1」以外の事象)及び臨床検査値の事前に規定した範囲を超える変動については、いずれかの投与群で4例以上に発現した事象は「Tier 2」の事象とし、群間差の95%信頼区間を算出した。いずれの投与群でも4例未満の事象は「Tier 3」の事象とし、M&N法を用いて投与群ごとの要約集計のみ行った。
※各薬剤の電子添文に記載されている最高用量の半量以下
| *FAS(Full Analysis Set): | 有効性評価に関する最大の解析対象集団/基礎試験用の治験薬を1回以上投与され、ベースライン値及び治験薬投与後の測定値を1つ以上有するすべての患者 |
安全性 (安全性解析対象集団、臨床検査値異常含む、レスキュー治療開始後データ除く)
本試験における副作用はマリゼブ®0.25mg/週 群113例中7例(6.2%)、1mg/週 群115例中6例(5.2%)、3mg/週 群114例中9例(7.9%)、10mg/週
群115例中9例(7.8%)、25mg/週 群114例中8例(7.0%)、プラセボ群113例中9例(8.0%)に認められた。発現率1%以上の主な副作用は、マリゼブ®0.25mg/週
群において高血糖が2例(1.8%)、10mg/週 群で血中尿酸増加が2例(1.7%)、25mg/週 群において頭痛が2例(1.8%)、プラセボ群において高血糖が2例(1.8%)であった。
重篤な副作用はマリゼブ®25mg/週 群において急性腎盂腎炎が1例であった。
投与中止に至った副作用は、マリゼブ®3mg/週 群において、血中クレアチンホスホキナーゼ増加が1例、25mg/週 群において急性腎盂腎炎、膀胱炎、尿管炎が各1例であった。
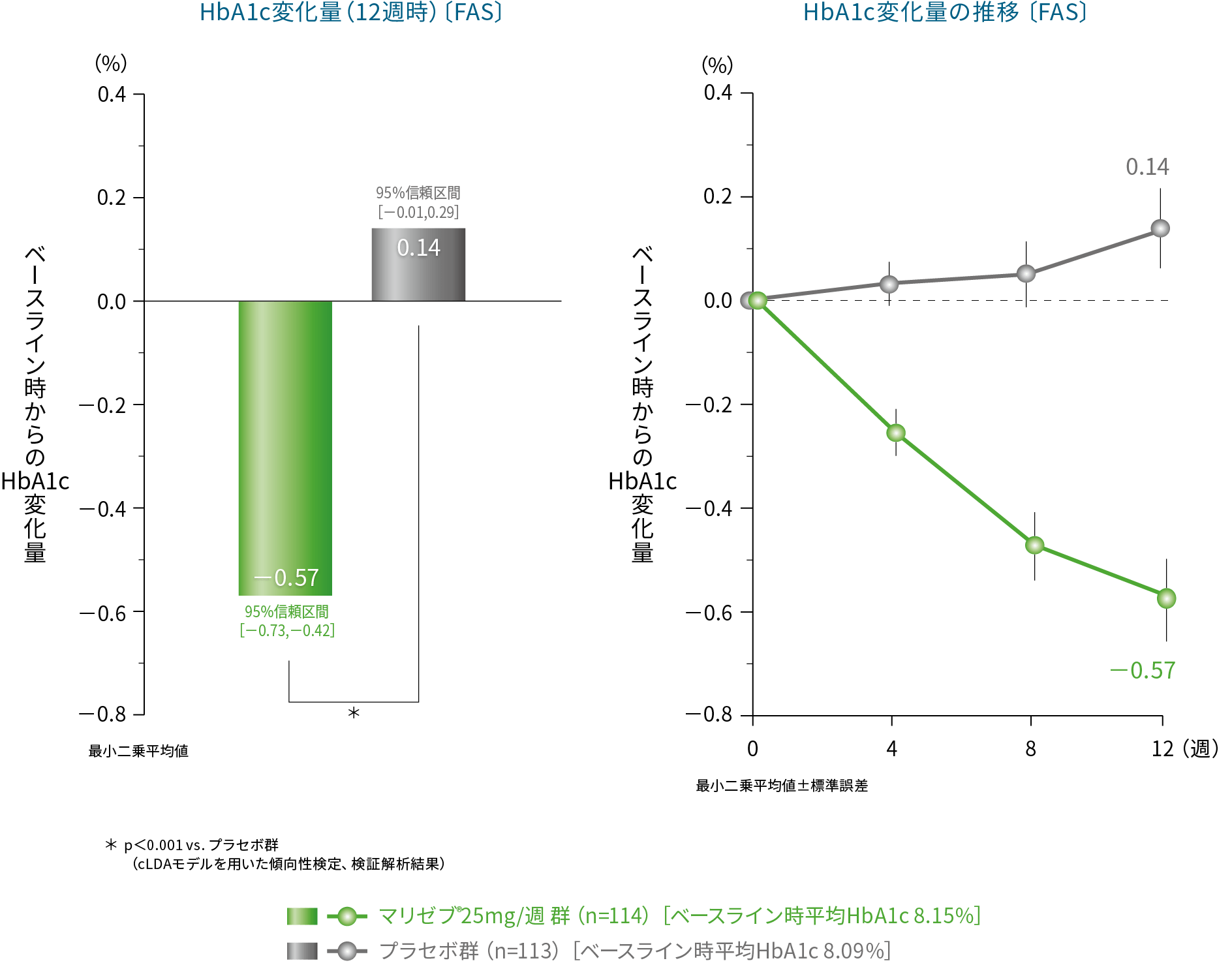
(1)投与12週時におけるHbA1cのベースライン時からの変化量1) 主要評価項目(検証的解析結果)
投与12週時におけるベースライン時からのHbA1c変化量〔最小二乗平均値[95%信頼区間]〕は、マリゼブ® 25mg/週 群では-0.57%[-0.73、-0.42]、プラセボ群では0.14%[-0.01、0.29]であり、マリゼブ® 25mg/週 群は、プラセボ群に対して有意な低下が検証された(p<0.001 vs. プラセボ群、cLDAモデルを用いた傾向性検定)。
cLDAモデル(constrained Longitudinal Data Analysis): 制約つき経時データ解析
1)社内資料(承認時評価資料):国際共同後期第Ⅱ相用量設定試験(P006-00)(2015年9月28日承認、CTD2.7.6.3.1)
-
6. 用法及び用量
- 通常、成人にはオマリグリプチンとして25mgを1週間に1回経口投与する。
2.第
Ⅲ相無作為二重盲検比較試験+非盲検延長試験
(単剤投与による非劣性試験)
2)
2)社内資料(承認時評価資料):第Ⅲ相臨床試験(P020)(2015年9月28日承認、CTD2.7.6.3.3)
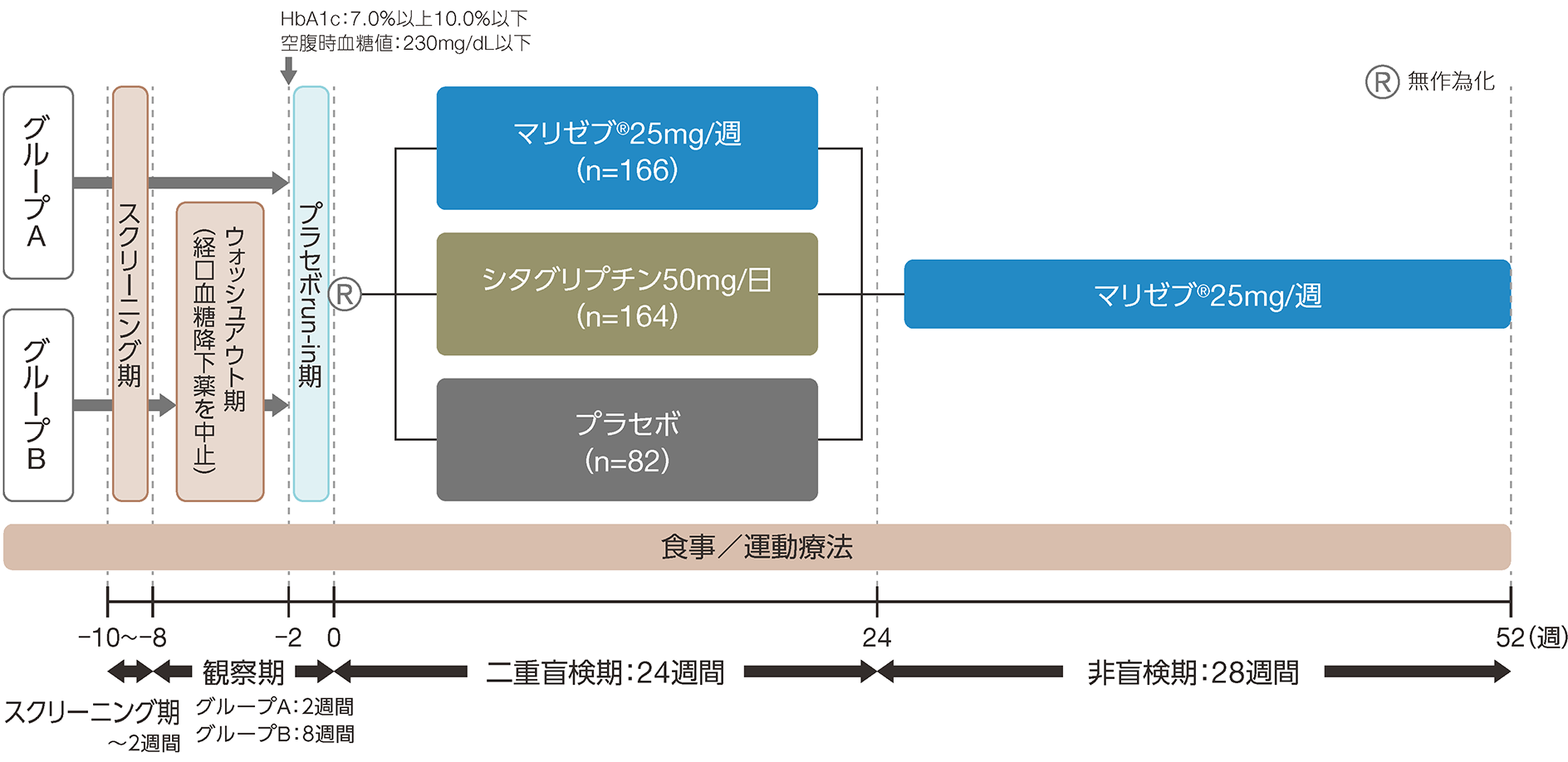
試験概要
- 目的:
- 食事/運動療法で十分な血糖管理が得られない日本人2型糖尿病患者に対し、マリゼブ®25mg/週を24週投与したときの有効性と安全性、ならびに52週投与したときの安全性と忍容性を検討する。
- 対象:
- 食事/運動療法で血糖管理不十分な2型糖尿病患者414例
- 経口血糖降下薬を6週間未投与でHbA1c7.0%以上10.0%以下の患者(グループA:観察期2週間)
- 経口血糖降下薬を単剤投与中でHbA1c6.5%以上9.0%以下の患者(グループB:観察期8週間)
[FAS *解析対象例数:マリゼブ®25mg/週 群166例、シタグリプチン50mg/日 群164例、プラセボ群82例]
- 試験デザイン:
- 無作為化、シタグリプチン及びプラセボ対照、並行群間、多施設共同、二重盲検試験及び引き続き実施される非盲検延長試験
- 投与方法:
- プラセボを単盲検下で2週間経口投与した後、無作為に割り付けた。二重盲検下でマリゼブ®25mg/週、シタグリプチン50mg/日またはプラセボを24週間経口投与した。
続いて、非盲検下でマリゼブ®25mg/週を週1回28週間経口投与した。
- 評価項目:
-
<主要評価項目> 投与24週時のHbA1cのベースライン時(投与0週時)からの変化量(検証的解析項目) <副次評価項目> 投与24週時の食後2時間血糖値、空腹時血糖値のベースライン時(投与0週時)からの変化量 <その他の有効性評価項目> 投与24週時のHbA1c<7.0%達成率、投与52週時のHbA1c、食後2時間血糖値、空腹時血糖値のベースライン時(投与0週時)からの変化量 <安全性評価項目> 有害事象、臨床検査値の事前に規定した範囲を超える変動、臨床検査値、12誘導心電図、バイタルサイン、体重 - 解析計画:
-
- 主要評価項目の投与24週時のHbA1cのベースライン時からの平均変化量において、マリゼブ®群のプラセボ群に対する優越性はcLDAモデルに基づいて検証された。マリゼブ® 25mg/週 群のプラセボ群に対する優越性が示された場合にマリゼブ® 25mg/週 群とシタグリプチン50mg/日 群の非劣性の検証を実施することとした。マリゼブ®群のシタグリプチン群に対する非劣性はcLDAモデルに基づき検証され、投与群間の最小二乗平均値の差(シタグリプチン-マリゼブ®)の95%信頼区間の上限値が0.3%を超えない場合に、マリゼブ®のシタグリプチンへの非劣性が検証されるものとした。また、投与24週時までのHbA1c変化量は最小二乗平均値±標準誤差で評価した。
- 副次評価項目も主要評価項目のHbA1c値の解析で用いたcLDAモデルと同様のモデルを用いて投与群ごとの投与24週時の食後2時間血糖値及び空腹時血糖値のベースライン時からの変化量を最小二乗平均値で評価した。また、マリゼブ®群とプラセボ群の変化量の有意差検定を実施した。
- その他の有効性評価項目はすべての投与群の治療期0週から治療期52週までのデータに対し、上記と同じcLDA モデルを用い、二重盲検期の投与群ごとに評価した。
- 安全性評価項目のうち、24週時は事前に規定した特に関心のある事象を「Tier 1」の事象(症候性低血糖症)とし、群間比較の統計的検定のp値及び95%信頼区間をM&N法を用いて算出した。群間比較は各実薬群とプラセボ群間で行われた。いずれかの投与群で4例が観測された事象は「Tier 2」の事象とし、それ以外の事象を「Tier 3」の事象とした。「Tier 2」の事象は、群間比較の群間差の点推定値を算出し、95%信頼区間をM&N法を用いて評価した。「Tier 3」の事象は投与群ごとの点推定値などの要約集計のみを行った。52週時においては、有害事象または臨床検査値における事前に規定した範囲を超える変動の発現率、及び連続値の安全性評価項目の要約統計量を、二重盲検期の投与群ごとに評価した。二重盲検期のマリゼブ®25mg/週 群は、本試験の全期間(52週間)の結果を報告した。二重盲検期のプラセボ群またはシタグリプチン50mg/日 群の結果は、マリゼブ®25mg/週 群が投与された28週間の結果を報告した。また、長期投与における体重変化量の推移は最小二乗平均値±標準誤差で評価した。
| *FAS(Full Analysis Set): | 有効性評価に関する最大の解析対象集団/基礎試験用の治験薬を1回以上投与され、ベースライン値及び治験薬投与後の測定値を1つ以上有するすべての患者 |
安全性 (安全性解析対象集団)(臨床検査値異常含む)
投与24週時:本試験における副作用は、マリゼブ®25mg/週 群166例中7例(4.2%)、シタグリプチン50mg/日
群164例中6例(3.7%)、プラセボ群82例中5例(6.1%)に認められた。発現率1%以上の主な副作用は、マリゼブ®25mg/週
群は、アラニンアミノトランスフェラーゼ増加2例(1.2%)、プラセボ群は腹部不快感、下痢、口内炎、低血糖症、皮膚炎が各1例(1.2%)であった。シタグリプチン50mg/日 群は認められなかった。
各群における重篤な副作用は認められなかった。
投与中止に至った副作用は、マリゼブ®25mg/週 群で上気道感染が1例、シタグリプチン50mg/日 群で悪心、流涎過多が各1例、プラセボ群で皮膚炎が1例であった。
各群において死亡例は報告されなかった。
投与52週時:副作用は、マリゼブ®25mg/週/マリゼブ®25mg/週 群
※1166例中13例(7.8%)、シタグリプチン50mg/日/
マリゼブ®25mg/週 群
※2161例中4例(2.5%)、プラセボ/マリゼブ®25mg/週 群
※280例中3例(3.8%)に認められた。発現率1%以上の主な副作用は、マリゼブ®25mg/週/マリゼブ®25mg/週 群
※1はアラニンアミノトランスフェラーゼ増加4例(2.4%)、血中ブドウ糖増加2例(1.2%)、グリコヘモグロビン増加2例(1.2%)、プラセボ/マリゼブ®25mg/週
群
※2は血中アルカリホスファターゼ増加、尿中血陽性、夜間頻尿、そう痒性皮疹が各1例(1.3%)であった。シタグリプチン50mg/日/マリゼブ®25mg/週
群
※2は認められなかった。
重篤な副作用は、マリゼブ®25mg/週/マリゼブ®25mg/週 群
※1で前立腺癌が1例であった。
投与中止に至った副作用は、マリゼブ®25mg/週/マリゼブ®25mg/週 群
※1で薬疹、血中ブドウ糖増加、グリコヘモグロビン増加が各1例、プラセボ/マリゼブ®25mg/週 群
※2でそう痒性皮疹が1例であった。
各群において死亡例は報告されなかった。
※2マリゼブ®25mg/週 28週間投与における副作用
●投与24週時
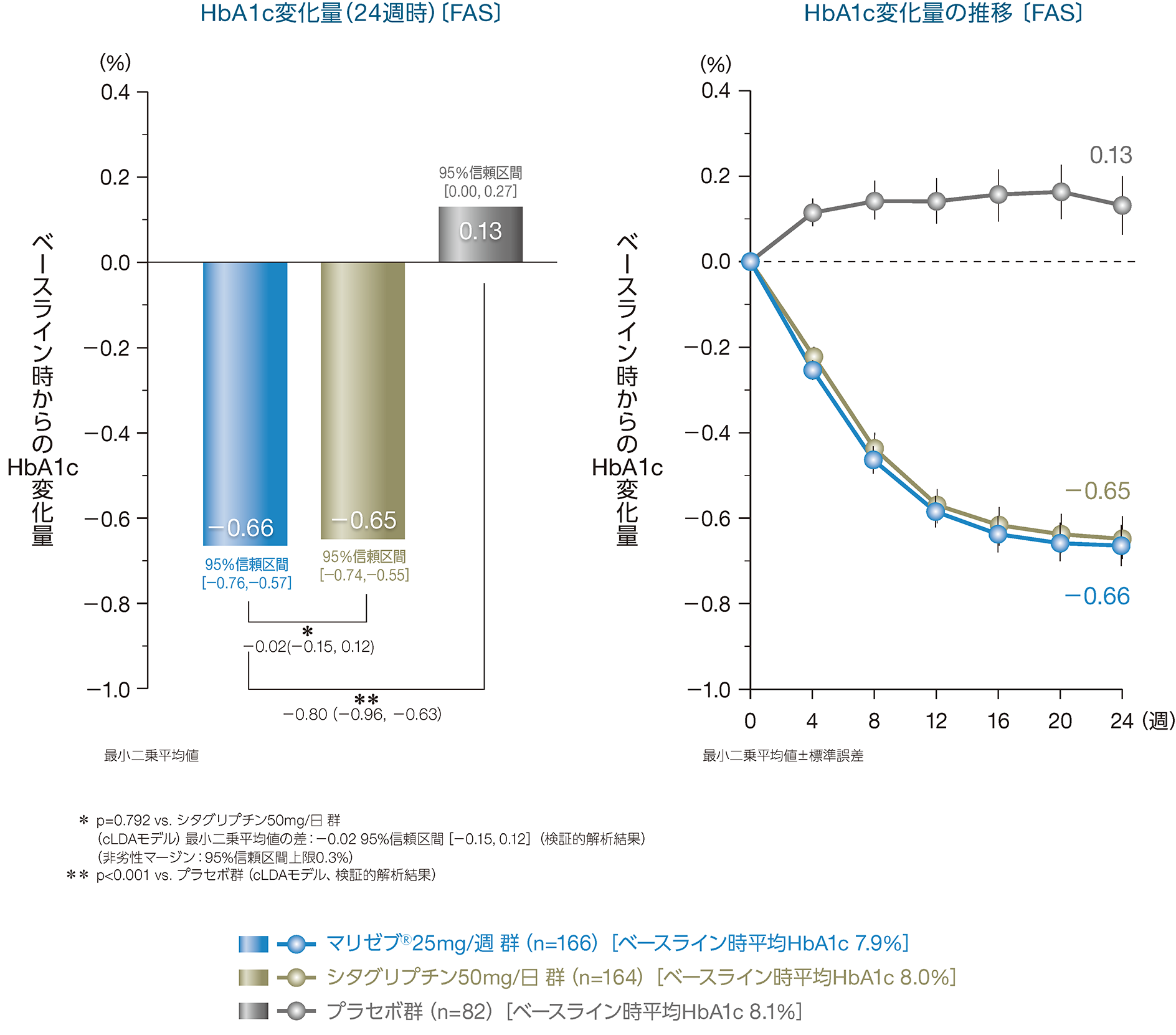
(1)投与12週時におけるHbA1cのベースライン時からの変化量2) 主要評価項目(検証的解析結果)
投与24週時におけるHbA1cのベースライン時からの変化量〔最小二乗平均値[95%信頼区間]〕は、マリゼブ®25mg/週
群では-0.66%[-0.76、-0.57]、シタグリプチン50mg/日
群では-0.65%[-0.74、-0.55]、プラセボ群では0.13%[0.00、0.27]であり、プラセボに対する優越性が検証されました(p<0.001 vs.プラセボ群、cLDAモデル)。
マリゼブ®25mg/週 群とシタグリプチン50mg/日
群のHbA1c変化量の最小二乗平均値の差は-0.02%[-0.15、0.12]であり、95%信頼区間の上限値が事前に設定した非劣性マージン(0.3%)を超えなかったことから、マリゼブ®25mg/週
群のシタグリプチン50mg/日 群に対する非劣性が検証されました(p=0.792 vs.シタグリプチン 50mg/日 群、cLDAモデル)。
cLDAモデル(constrained Longitudinal Data Analysis): 制約つき経時データ解析
2)社内資料(承認時評価資料):第Ⅲ相臨床試験(P020)(2015年9月28日承認、CTD2.7.6.3.3)
(2)食後2時間血糖値、空腹時血糖値のベースライン時からの変化量 3) 副次評価項目
投与23週の投与7日後のベースライン時からの食後2時間血糖値(2-hour PMG)及び空腹時血糖値(FPG)の変化量〔最小二乗平均値[95%信頼区間]〕は、マリゼブ®25mg/週 群ではそれぞれ-43.0mg/dL[-50.9、-35.1]及び-19.4mg/dL[−23.2、-15.6]でした。マリゼブ®25mg/週 群とプラセボ群との2-hour PMG及びFPGの最小二乗平均値の差は、それぞれ-34.5mg/dL[-46.9、-22.1]及び-11.4mg/dL[-17.6、-5.3]であり、いずれもマリゼブ®25mg/週 群はプラセボ群に対して有意に低値を示しました(いずれもp<0.001(名目上のp値) vs.プラセボ群、cLDAモデル)。

| *PP(Per Protocol): | 治験実施計画書に適合した対象集団(投与7日後に血糖値を測定した対象集団) |
| cLDAモデル(constrained Longitudinal Data Analysis): | 制約つき経時データ解析 |
3)社内資料(承認時評価資料):第Ⅲ相臨床試験(P020)(2015年9月28日承認、CTD2.7.3.4.2.1)
(3) 投与24週時のHbA1c7.0%未満達成率 2) その他の有効性評価項目
投与24週時にHbA1c7.0%未満を達成した患者は、マリゼブ®25mg/週 群において166例中78例(47%)でした。
2)社内資料(承認時評価資料):第Ⅲ相臨床試験(P020)(2015年9月28日承認、CTD2.7.6.3.3)
●投与52週時
(1)長期投与におけるHbA1c変化量の推移 2) その他の有効性評価項目
投与52週時におけるベースライン時からのHbA1c変化量〔最小二乗平均値[95%信頼区間]〕は、-0.37%[-0.47、-0.26]でした。
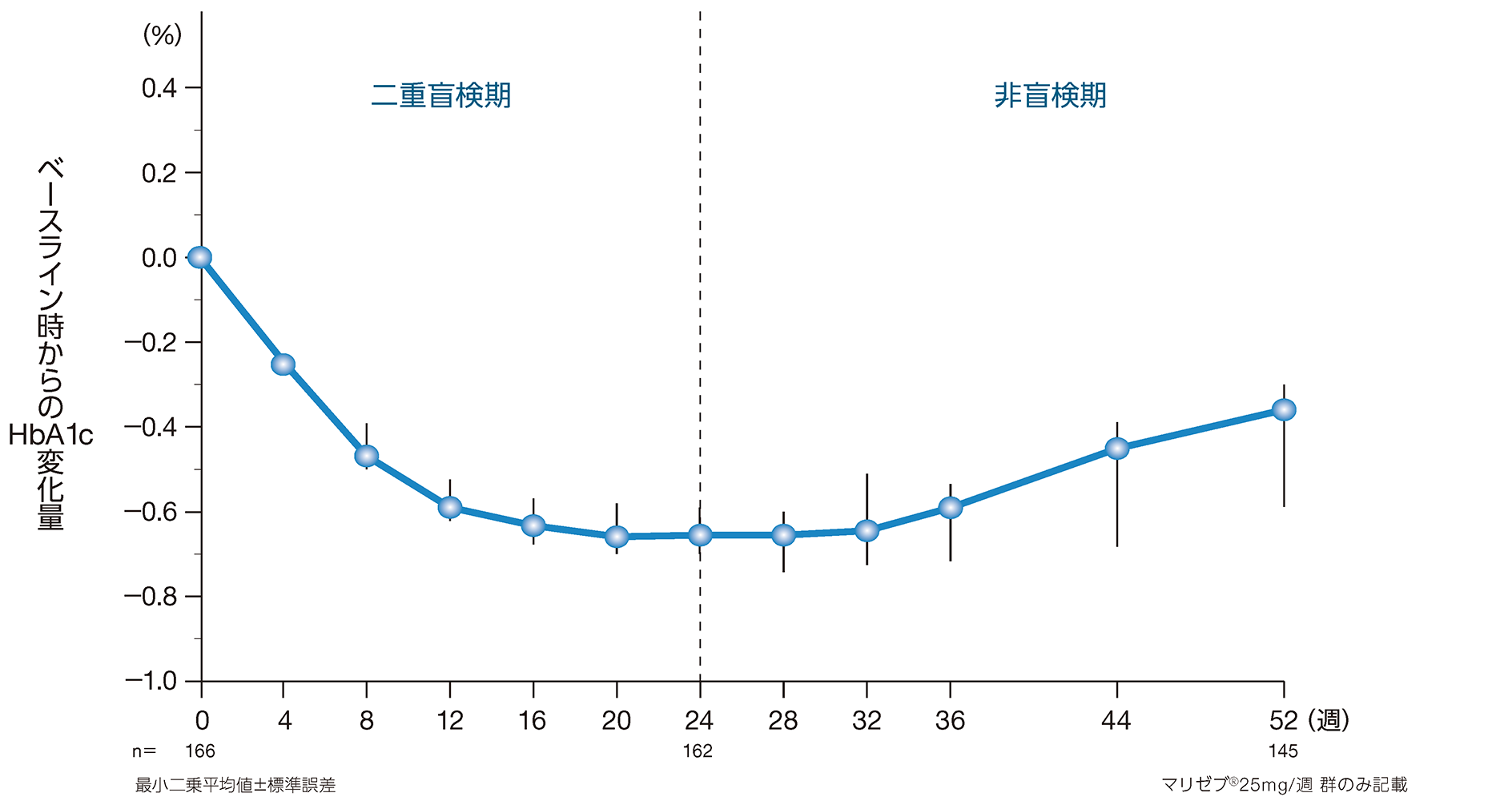
2)社内資料(承認時評価資料):第Ⅲ相臨床試験(P020)(2015年9月28日承認、CTD2.7.6.3.3)
(2)長期投与における体重変化量の推移(52週間投与) 2) 安全性評価項目
投与52週時におけるベースライン時からの体重変化量〔最小二乗平均値[95%信頼区間]〕は、マリゼブ® 25mg/週群では0.37kg[0.04、0.71]でした。
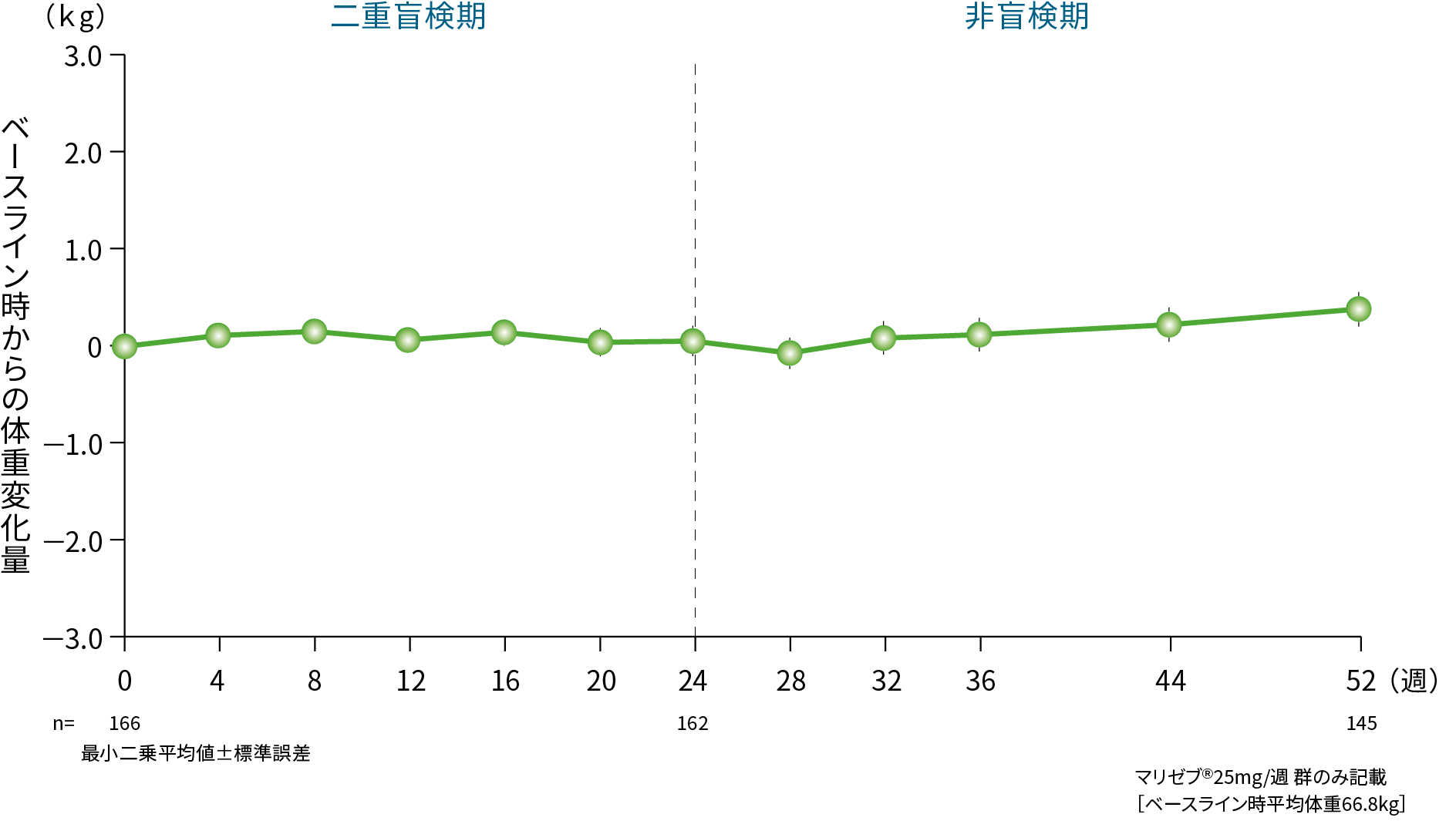
2)社内資料(承認時評価資料):第Ⅲ相臨床試験(P020)(2015年9月28日承認、CTD2.7.6.3.3)
-
8. 重要な基本的注意(抜粋)
- 8.2本剤投与中は、血糖を定期的に検査するとともに、経過を十分に観察し、常に投与継続の必要性について注意を払うこと。本剤を3ヵ月投与しても効果が不十分な場合、より適切と考えられる治療への変更を考慮すること。
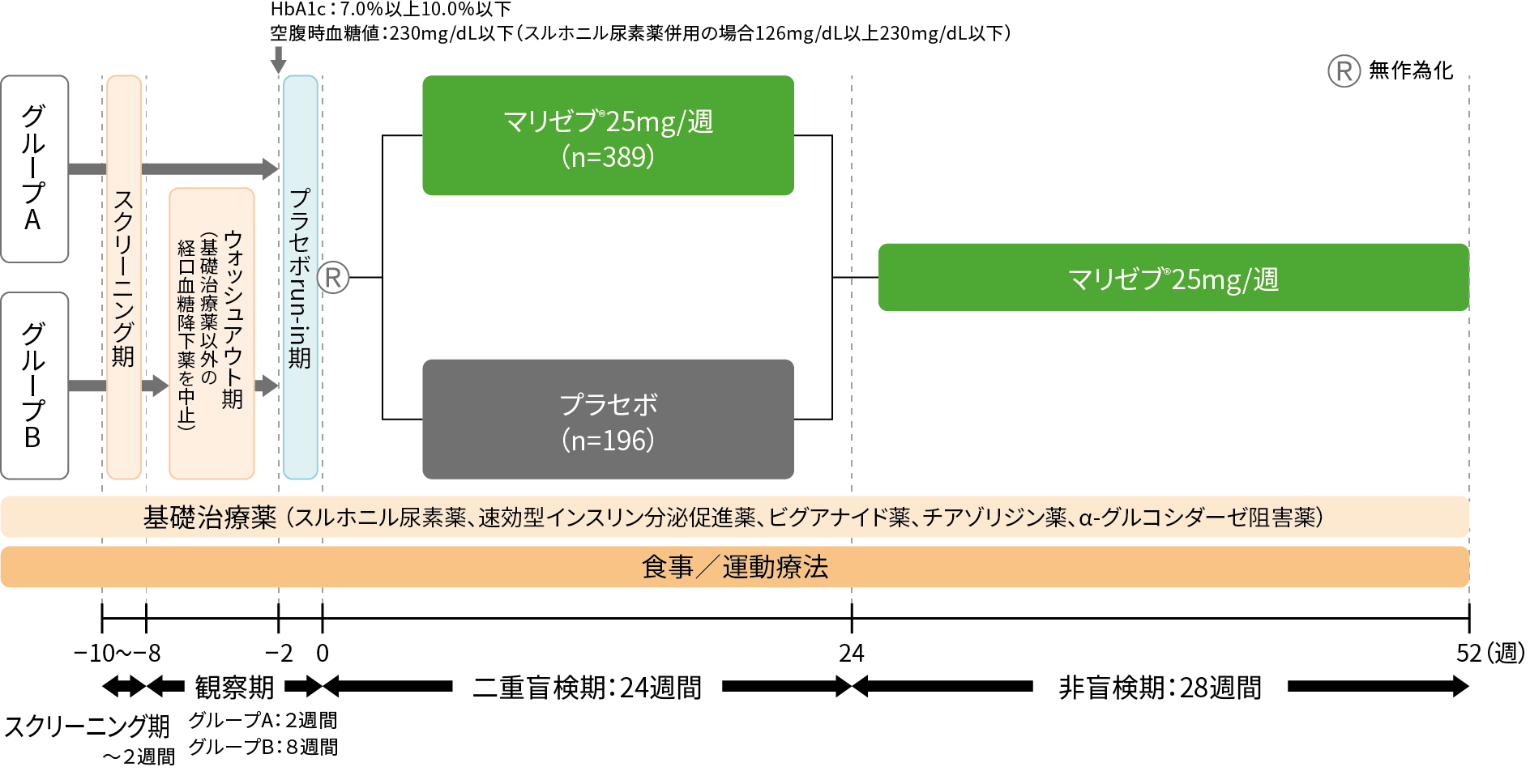
3.第 Ⅲ相無作為二重盲検比較試験+非盲検延長試験(他剤との併用試験) 4)
4)社内資料(承認時評価資料):第Ⅲ相臨床試験(P015)(2015年9月28日承認、CTD2.7.6.3.4)
試験概要
- 目的:
- 食事/運動療法に加え経口糖尿病治療薬単剤治療で十分な血糖管理が得られない日本人2型糖尿病患者に対するマリゼブ®25mg/週 追加投与の安全性、忍容性及び有効性を検討する。
- 対象:
- 食事/運動療法に加え経口糖尿病治療薬(基礎治療薬)単剤治療で血糖管理不十分な2型糖尿病患者585例
- 基礎治療薬 ※1,2以外の経口糖尿病治療薬を6週間未投与でHbA1c7.0%以上10.0%以下の患者(グループA:観察期2週間)
- 基礎治療薬 ※1,3以外の経口糖尿病治療薬を1剤投与中でHbA1c6.5%以上9.0%以下の患者(グループB:観察期8週間)
[FAS *解析対象例数:スルホニル尿素薬189例、速効型インスリン分泌促進薬99例、ビグアナイド薬99例、チアゾリジン薬99例、α-グルコシダーゼ阻害薬99例]
- 試験デザイン:
- 無作為化、プラセボ対照、並行群間、多施設共同、二重盲検試験及び引き続き実施される非盲検延長試験
- 投与方法:
- プラセボを単盲検下で2週間経口投与した後、基礎治療薬
※1ごとにマリゼブ®25mg/週
群またはプラセボ群に無作為に割り付けた。まず、二重盲検下でマリゼブ®25mg/週またはプラセボを週1回、24週間経口投与を行い、続いて非盲検下でマリゼブ®25mg/週を週1回28週間経口投与した。投与開始4週時以降、血糖値が基準に達しない場合は
①基礎治療薬 ※1の増量、②メトホルミンまたはグリメピリド追加、によるレスキュー治療を行った。基礎治療薬 ※1はレスキュー基準に該当しない限り、一定かつ電子添文に従った適切な用法及び用量での服用を継続した。
- 評価項目:
-
<主要評価項目> 投与24週時及び52週時の安全性(有害事象、臨床検査値の事前に規定した範囲を超える変動、臨床検査値、12誘導心電図、バイタルサイン、体重) <副次評価項目> 投与24週及び52週時のHbA1cのベースライン時(投与0週時)からの変化量 <安全性評価項目> 症候性低血糖症、有害事象、臨床検査値、12誘導心電図、バイタルサイン、体重 - 解析計画:
-
- 主要評価項目のうち、24週時は事前に規定した特に関心のある事象を「Tier 1」の事象(症候性低血糖症)とし、群間比較の統計的検定のp値及び95%信頼区間をM&N法を用いて算出した。群間比較は各実薬群とプラセボ群間で行われた。いずれかの投与群で4例が観測された事象は「Tier 2」の事象とし、それ以外の事象を「Tier 3」の事象とした。「Tier 2」の事象は、群間比較の群間差の点推定値を算出し、95%信頼区間をM&N法を用いて評価した。「Tier 3」の事象は投与群ごとの点推定値などの要約集計のみを行った。52週時においては、有害事象または臨床検査値及び12誘導心電図における事前に規定した範囲を超える変動の発現率、及び連続値の安全性評価項目の要約統計量を、二重盲検期の投与群ごとに評価した。レスキュー後のデータを除いた解析を主たるアプローチとして事前定義した。死亡、その他の重篤な有害事象、ならびに中止に至った有害事象は、レスキュー後のデータも示した。
- 副次評価項目のうち、24週時は反復測定されたデータ間の相関をモデル化するために無構造型の共分散行列を用いた。治療期24週時のベースラインからのHbA1c値の平均変化量におけるマリゼブ®25mg/週 群のプラセボ群に対する優越性については、本cLDAモデルを用いて解析を行った。52週時においては、治療期24週時に適用したcLDAモデルでは収束しなかったため、治療期24週時に適用したcLDAモデルから基礎治療薬 ※1に関連した項をすべて除き、基礎治療薬ごとにcLDAモデルを適用して解析した。
- 安全性及び忍容性は、基礎治療薬ごとに有害事象、臨床検査値、12誘導心電図、バイタルサイン及び体重を含むすべての安全性評価項目を用いて、臨床的な観点から評価した。適宜、基礎治療薬を併合した集計も行った。臨床検査値、12誘導心電図、バイタルサイン及び体重のベースラインからの変化量も「Tier 3」の評価項目とした。臨床検査値、12誘導心電図及びバイタルサインの解析には、治験薬投与後の測定値を1つ以上有する被験者を含めた。ベースラインからの変化量の解析には、ベースライン値も必要とした。治療期52週時の解析では、プラセボ/マリゼブ®群のベースライン値は、マリゼブ®投与前の治療期24週時の測定値とした。ベースラインからの変化量の解析では、欠測値は補完しなかった。
※2チアゾリジン薬は14週間以上、その他の経口血糖降下薬は10週間以上投与
※3チアゾリジン薬は8週間以上、その他の経口血糖降下薬は4週間以上投与
| *FAS(Full Analysis Set): | 有効性評価に関する最大の解析対象集団/基礎試験用の治験薬を1回以上投与され、ベースライン値及び治験薬投与後の測定値を1つ以上有するすべての患者 |
安全性5) 主要評価項目 (安全性解析対象集団)(臨床検査値異常含む、レスキュー治療開始後データ除く)
<全集団>
| マリゼブ®25mg/週 群
(n=389) |
プラセボ群
(n=196) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | 21(5.4%) | 8(4.1%) |
| 心室性期外収縮 | - | 1(0.5%) | |
| 腹部不快感 | 1(0.3%) | - | |
| 便秘 | 3(0.8%) | 1(0.5%) | |
| 下痢 | 1(0.3%) | 1(0.5%) | |
| 悪心 | 2(0.5%) | - | |
| 嘔吐 | 1(0.3%) | - | |
| 胸痛 | 1(0.3%) | - | |
| アラニンアミノトランスフェラーゼ増加 | 2(0.5%) | - | |
| アスパラギン酸アミノトランスフェラーゼ増加 | 1(0.3%) | - | |
| 血中クレアチンホスホキナーゼ増加 | 2(0.5%) | - | |
| 高血糖 | - | 1(0.5%) | |
| 低血糖症 | 7(1.8%) | 3(1.5%) | |
| 失神寸前の状態 | - | 1(0.5%) | |
| 卵巣嚢胞 | 1(0.3%) | - | |
| 湿疹 | 1(0.3%) | - | |
| 紫斑 | 1(0.3%) | - | |
| マリゼブ®25mg/週/
マリゼブ®25mg/週 群 (n=389) |
プラセボ/
マリゼブ®25mg/週 群 (n=191) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | 24(6.2%) | 8(4.2%) |
| 上室性期外収縮 | 1(0.3%) | - | |
| 発作性頻脈 | - | 1(0.5%) | |
| 腹部不快感 | 1(0.3%) | 1(0.5%) | |
| 便秘 | 5(1.3%) | - | |
| 下痢 | 1(0.3%) | - | |
| 悪心 | 2(0.5%) | - | |
| 嘔吐 | 1(0.3%) | - | |
| 胸痛 | 1(0.3%) | - | |
| アラニンアミノトランスフェラーゼ増加 | 1(0.3%) | - | |
| アスパラギン酸アミノトランスフェラーゼ増加 | 1(0.3%) | - | |
| 血中クレアチニン増加 | 1(0.3%) | - | |
| 低血糖症 | 10(2.6%) | 4(2.1%) | |
| 不規則月経 | - | 1(0.5%) | |
| 卵巣嚢胞 | 1(0.3%) | - | |
| 湿疹 | 1(0.3%) | 2(1.0%) | |
| 多形紅斑 | 1(0.3%) | - | |
| 紫斑 | 1(0.3%) | - | |
二重盲検期(24週)、非盲検期(52週)における死亡例を含む重篤な副作用はなかった。
投与中止に至った副作用は、以下の通りであった。
- 速効型インスリン分泌促進薬併用
二重盲検期(24週)におけるマリゼブ 25mg/週 群で腹部不快感 1例
<スルホニル尿素薬併用>
| マリゼブ®25mg/週 群
(n=126) |
プラセボ群
(n=63) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | 9(7.1%) | 5(7.9%) |
| 便秘 | 2(1.6%) | 1(1.6%) | |
| 下痢 | - | 1(1.6%) | |
| 悪心 | 1(0.8%) | - | |
| 血中クレアチンホスホキナーゼ増加 | 1(0.8%) | - | |
| 低血糖症 | 4(3.2%) | 3(4.8%) | |
| 卵巣嚢胞 | 1(0.8%) | - | |
| マリゼブ®25mg/週/
マリゼブ®25mg/週 群 (n=126) |
プラセボ/
マリゼブ®25mg/週 群 (n=62) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | 11(8.7%) | 5(8.1%) |
| 上室性期外収縮 | 1(0.8%) | - | |
| 便秘 | 3(2.4%) | - | |
| 悪心 | 1(0.8%) | - | |
| 低血糖症 | 5(4.0%) | 4(6.5%) | |
| 卵巣嚢胞 | 1(0.8%) | - | |
| 湿疹 | - | 1(1.6%) | |
| 多形紅斑 | 1(0.8%) | - | |
各群において二重盲検期(24週)、非盲検期(52週)における死亡例を含む重篤な副作用、投与中止に至った副作用はなかった。
<速効型インスリン分泌促進薬併用>
| マリゼブ®25mg/週 群 (n=65) |
プラセボ群 (n=34) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | 5(7.7%) | 2(5.9%) |
| 心室性期外収縮 | - | 1(2.9%) | |
| 腹部不快感 | 1(1.5%) | - | |
| 便秘 | 1(1.5%) | - | |
| 悪心 | 1(1.5%) | - | |
| 嘔吐 | 1(1.5%) | - | |
| 胸痛 | 1(1.5%) | - | |
| アラニンアミノトランスフェラーゼ増加 | 1(1.5%) | - | |
| 失神寸前の状態 | - | 1(2.9%) | |
| 湿疹 | 1(1.5%) | - | |
| マリゼブ®25mg/週/ マリゼブ®25mg/週 群 (n=65) |
プラセボ/ マリゼブ®25mg/週 群 (n=32) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | 7(10.8%) | 2(6.3%) |
| 発作性頻脈 | - | 1(3.1%) | |
| 腹部不快感 | 1(1.5%) | 1(3.1%) | |
| 便秘 | 2(3.1%) | - | |
| 悪心 | 1(1.5%) | - | |
| 嘔吐 | 1(1.5%) | - | |
| 胸痛 | 1(1.5%) | - | |
| アラニンアミノトランスフェラーゼ増加 | 1(1.5%) | - | |
| アスパラギン酸アミノトランスフェラーゼ増加 | 1(1.5%) | - | |
| 低血糖症 | 1(1.5%) | - | |
| 不規則月経 | - | 1(3.1%) | |
| 湿疹 | 1(1.5%) | - | |
各群において二重盲検期(24週)、非盲検期(52週)における死亡例を含む重篤な副作用はなかった。
投与中止に至った副作用は、二重盲検期(24週)におけるマリゼブ®25mg/週 群で腹部不快感が1例であった。
<ビグアナイド薬併用>
| マリゼブ®25mg/週 群 (n=66) |
プラセボ群 (n=33) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | 3(4.5%) | - |
| アラニンアミノトランスフェラーゼ増加 | 1(1.5%) | - | |
| アスパラギン酸アミノトランスフェラーゼ増加 | 1(1.5%) | - | |
| 低血糖症 | 2(3.0%) | - | |
| マリゼブ®25mg/週/ マリゼブ®25mg/週 群 (n=66) |
プラセボ/ マリゼブ®25mg/週 群 (n=33) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | 2(3.0%) | - |
| 低血糖症 | 2(3.0%) | - | |
各群において二重盲検期(24週)、非盲検期(52週)における死亡例を含む重篤な副作用、投与中止に至った副作用はなかった。
<チアゾリジン薬併用>
| マリゼブ®25mg/週 群 (n=65) |
プラセボ群 (n=34) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | 4(6.2%) | - |
| 下痢 | 1(1.5%) | - | |
| 血中クレアチンホスホキナーゼ増加 | 1(1.5%) | - | |
| 低血糖症 | 1(1.5%) | - | |
| 紫斑 | 1(1.5%) | - | |
| マリゼブ®25mg/週/ マリゼブ®25mg/週 群 (n=65) |
プラセボ/ マリゼブ®25mg/週 群 (n=34) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | 4(6.2%) | - |
| 下痢 | 1(1.5%) | - | |
| 血中クレアチニン増加 | 1(1.5%) | - | |
| 低血糖症 | 2(3.1%) | - | |
| 紫斑 | 1(1.5%) | - | |
二重盲検期(24週)、非盲検期(52週)における死亡例を含む重篤な副作用、投与中止に至った副作用はなかった。
<αグルコシダーゼ阻害薬併用>
| マリゼブ®25mg/週 群 (n=67) |
プラセボ群 (n=32) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | - | 1(3.1%) |
| 高血糖 | - | 1(3.1%) | |
| マリゼブ®25mg/週/ マリゼブ®25mg/週 群 (n=67) |
プラセボ/ マリゼブ®25mg/週 群 (n=30) |
||
|---|---|---|---|
| 副作用 | 発現例数(発現率) | - | 1(3.3%) |
| 湿疹 | - | 1(3.3%) | |
各群において二重盲検期(24週)、非盲検期(52週)における死亡例を含む重篤な副作用、投与中止に至った副作用はなかった。
5)社内資料(承認時評価資料):第Ⅲ相臨床試験(P015)(2015年9月28日承認、CTD5.3.5.1.4)
●投与24週時
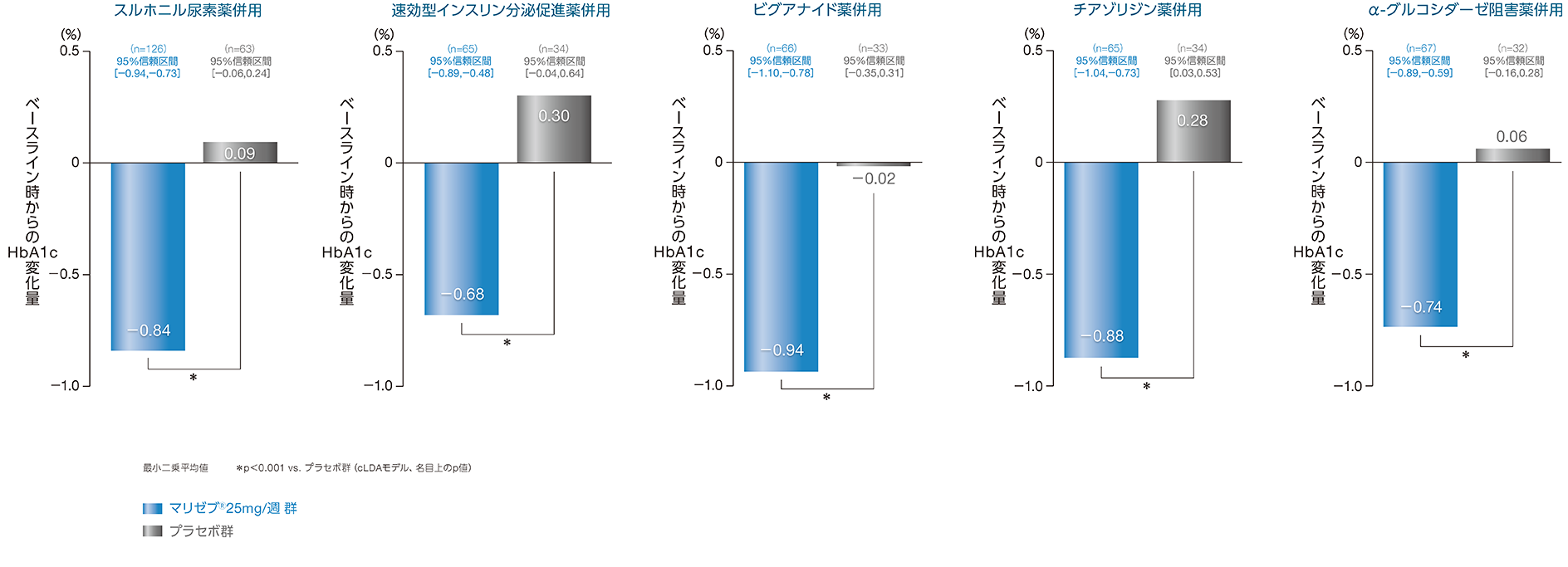
(1)HbA1c変化量4) 副次評価項目
マリゼブ®25mg/週を基礎治療薬(スルホニル尿素薬、速効型インスリン分泌促進薬、ビグアナイド薬、チアゾリジン薬、α-グルコシダーゼ阻害薬)に追加投与した結果、投与24週時におけるマリゼブ®
25mg/週
群のベースライン時からのHbA1c変化量〔最小二乗平均値[95%信頼区間]〕は、それぞれ-0.84%[-0.94、-0.73]、-0.68%[-0.89、-0.48]、-0.94%[-1.10、-0.78]、-0.88%[-1.04、-0.73]、-0.74%[-0.89、-0.59]でした。
マリゼブ®25mg/週
群とプラセボ群のHbA1c変化量の最小二乗平均値の差は、それぞれ-0.93%[-1.10、-0.75]、-0.98%[-1.37、-0.60]、-0.92%[-1.29、-0.56]、-1.16%[-1.45、-0.88]、-0.80%[-1.06、-0.54]であり、いずれもマリゼブ®25mg/週
群はプラセボ群に対して有意に低値を示しました(いずれもp<0.001(名目上のp値) vs.プラセボ群、cLDAモデル)。
cLDAモデル(constrained Longitudinal Data Analysis): 制約つき経時データ解析
4)社内資料(承認時評価資料):第Ⅲ相臨床試験(P015)(2015年9月28日承認、CTD2.7.6.3.4)
-
8.重要な基本的注意(抜粋)
- 8.1本剤の使用にあたっては、患者に対し低血糖症状及びその対処方法について十分説明すること。[9.1.1、11.1.1 参照]
- 8.2本剤投与中は、血糖を定期的に検査するとともに、経過を十分に観察し、常に投与継続の必要性について注意を払うこと。本剤を3ヵ月投与しても効果が不十分な場合、より適切と考えられる治療への変更を考慮すること。
- 8.5本剤とGLP-1受容体作動薬はいずれもGLP-1受容体を介した血糖降下作用を有している。両剤を併用した際の臨床試験成績はなく、有効性及び安全性は確認されていない。
-
10.相互作用(抜粋)
- 本剤は主に腎臓から未変化体として排泄され、排泄には糸球体濾過及び再吸収が関与する。[16.5.1-16.5.4参照]
-
10.2 併用注意(併用に注意すること)
糖尿病用薬:インスリン製剤、スルホニルウレア剤、チアゾリジン系薬剤、ビグアナイド系薬剤、α-グルコシダーゼ阻害剤、速効型インスリン分泌促進薬、GLP-1受容体作動薬、SGLT2阻害剤等[11.1.1参照]
血糖降下作用を増強する薬剤:β-遮断薬、サリチル酸剤、モノアミン酸化酵素阻害剤等
血糖降下作用を減弱する薬剤:アドレナリン、副腎皮質ホルモン、甲状腺ホルモン等
